-
We theoretically and experimentally investigate low-Reynolds-number
propulsion of geometrically achiral planar objects that possess a dipole moment
and that are driven by a rotating magnetic field. Symmetry considerations
(involving parity, $\widehat{P}$, and charge conjugation, $\widehat{C}$)
establish correspondence between propulsive states depending on orientation of
the dipolar moment. Although basic symmetry arguments do not forbid individual
symmetric objects to efficiently propel due to spontaneous symmetry breaking,
they suggest that the average ensemble velocity vanishes. Some additional
arguments show, however, that highly symmetrical ($\widehat{P}$-even) objects
exhibit no net propulsion while individual less symmetrical
($\widehat{C}\widehat{P}$-even) propellers do propel. Particular magnetization
orientation, rendering the shape $\widehat{C}\widehat{P}$-odd, yields
unidirectional motion typically associated with chiral structures, such as
helices. If instead of a structure with a permanent dipole we consider a
polarizable object, some of the arguments have to be modified. For instance, we
demonstrate a truly achiral ($\widehat{P}$- and $\widehat{C}\widehat{P}$-even)
planar shape with an induced electric dipole that can propel by
electro-rotation. We thereby show that chirality is not essential for
propulsion due to rotation-translation coupling at low Reynolds number.
-
Active particles disturb the fluid around them as force dipoles, or
stresslets, which govern their collective dynamics. Unlike swimming speeds, the
stresslets of active particles are rarely determined due to the lack of a
suitable theoretical framework for arbitrary geometry. We propose a general
method, based on the reciprocal theorem of Stokes flows, to compute stresslets
as integrals of the velocities on the particle's surface, which we illustrate
for spheroidal chemically-active particles. Our method will allow tuning the
stresslet of artificial swimmers and tailoring their collective motion in
complex environments.
-
Interpenetrated Polymer Network (IPN) microgels of PNIPAM and PAAc have been
investigated and the experimental data have been compared with theoretical
models from the Flory-Rehner theory. We confirm that the swelling behavior of
PNIPAM microgels is well described by this theory by considering the second
order approximation for the volume fraction $\phi$ dependence of the Flory
parameter $\chi(\phi)$. Indeed the Volume-Phase Transition (VPT) of the
PNIPAM-PAAc IPN microgel at neutral conditions and in D$_2$O solvents can be
well described only considering a third-order approximation. Interestingly we
empirically find that sharper is the transition higher is the order of the
$\chi(\phi)$ relation which has to be considered. Moreover the VPT can be
experimentally controlled by tuning the polymer/solvent interactions through pH
and solvent allowing to directly modify the delicate balance between energetic
and entropic contributions and to explore the swelling behavior in a wide range
of environmental conditions. In particular we find that the most advantageous
condition for swelling is in water at acidic pH.
-
Microgel suspensions of Interpenetrated Polymer Network (IPN) of PNIPAM and
PAAc in D$_2$O, have been investigated through dynamic light scattering as a
function of temperature, pH and concentration across the Volume Phase
Transition (VPT). The dynamics of the system is slowed down under H/D isotopic
substitution due to the different balance between polymer/polymer and
polymer/solvent interactions suggesting the crucial role played by H-bondings.
The swelling behavior, reduced with respect to PNIPAM and water, has been
described by the Flory-Rehner theory, tested for PNIPAM microgel and
successfully expanded to higher order for IPN microgels. Moreover the
concentration dependence of the relaxation time at neutral pH has highlighted
two different routes to approach the glass transition: Arrhenius and
super-Arrhenius (Vogel Fulcher Tammann) respectively below and above the VPT
and a fragility plot has been derived. Fragility can be tuned by changing
temperature: across the VPT particles undergo a transition from soft-strong to
stiff-fragile.
-
Microgels are colloidal-scale particles individually made of crosslinked
polymer networks that can swell and deswell in response to external stimuli,
such as changes to temperature or pH. Despite a large amount of experimental
activities on microgels, a proper theoretical description based on individual
particle properties is still missing due to the complexity of the particles. To
go one step further, here we propose a novel methodology to assemble realistic
microgel particles "in silico". We exploit the self-assembly of a binary
mixture composed of tetravalent (crosslinkers) and bivalent (monomer beads)
patchy particles under spherical confinement in order to produce fully-bonded
networks. The resulting structure is then used to generate the initial microgel
configuration, which is subsequently simulated with a bead-spring model
complemented by a temperature-induced hydrophobic attraction. To validate our
assembly protocol we focus on a small microgel test-case and show that we can
reproduce the experimental swelling curve by appropriately tuning the confining
sphere radius, something that would not be possible with less sophisticated
assembly methodologies, e.g. in the case of networks generated from an
underlying crystal structure. We further investigate the structure (in
reciprocal and real space) and the swelling curves of microgels as a function
of temperature, finding that our results are well described by the widely-used
fuzzy sphere model. This is a first step toward a realistic modelling of
microgel particles, which will pave the way for a careful assessment of their
elastic properties and effective interactions.
-
Effective colloid-colloid interactions can be tailored through the addition
of a complex cosolute. Here we investigate the case of a cosolute made by
self-assembling patchy particles. Depending on the valence, these particles can
form either polymer chains or branched structures. We numerically calculate the
effective potential $V_{eff}$ between two colloids immersed in a suspension of
reversible patchy particles, exploring a wide region of the cosolute phase
diagram and the role of valence. In addition to well-known excluded volume and
depletion effects, we find that, under appropriate conditions, $V_{eff}$ is
completely attractive but shows an oscillatory character. In the case of
polymerizing cosolute, this results from the fact that chains are efficiently
confined by the colloids through the onset of local order. This argument is
then generalized to the case of particles with higher valence, under the
condition that they are still able to maintain a fully bonded organization upon
confinement. The resulting effective potentials are relevant for understanding
the behavior of complex mixtures in crowded environments, but may also be
exploited for tuning colloidal self-assembly at preferred target distances in
order to build desired superstructures.
-
We develop topological methods for characterizing the relationship between
polymer chain entanglement and bulk viscoelastic responses. We introduce
generalized Linking Number and Writhe characteristics that are applicable to
open linear chains. We investigate the rheology of polymeric chains entangled
into weaves with varying topologies and levels of chain density. To investigate
viscoelastic responses, we perform non-equilibrium molecular simulations over a
range of frequencies using sheared Lees-Edwards boundary conditions. We show
how our topological characteristics can be used to capture key features of the
polymer entanglements related to the viscoelastic responses. We find there is a
linear relation over a significant range of frequencies between the mean
absolute Writhe $Wr$ and the Loss Tangent $\tan(\delta)$. We also find an
approximate inverse linear relationship between the mean absolute Periodic
Linking Number $LK_P$ and the Loss Tangent $\tan(\delta)$. Our results show
some of the ways topological methods can be used to characterize chain
entanglements to better understand the origins of mechanical responses in
polymeric materials.
-
Turing patterns emerge from a spatially uniform state following a linear
instability driven by diffusion. Features of the eventual pattern (stabilized
by non-linearities) are already present in the initial unstable modes. On a
uniform flat surface or perfect sphere, the unstable modes and final patterns
are degenerate, reflecting translational/rotational symmetry. This symmetry can
be broken, e.g. by a bump on a flat substrate or by deforming a sphere. As the
diffusion operator on a two dimensional manifold depends on the underlying
curvature, the degeneracy of the initial unstable mode is similarly reduced.
Different shapes can pin different modes. We adapt methods of conformal mapping
and perturbation theory to analytically examine how bumps and ripples entrain
modes of the diffusion operator on cylinders and spheres. We confirm these
results numerically, and provide closed form expressions that describe how
non-uniformities in curvature pin diffusion-driven instabilities and the
resulting patterns.
-
The Percus-Yevick theory for monodisperse hard spheres gives very good
results for the pressure and structure factor of the system in a whole range of
densities that lie within the liquid phase. However, the equation seems to lead
to a very unacceptable result beyond that region. Namely, the Percus-Yevick
theory predicts a smooth behavior of the pressure that diverges only when the
volume fraction $\eta$ approaches unity. Thus, within the theory there seems to
be no indication for the termination of the liquid phase and the transition to
a solid or to a glass. In the present article we study the Percus-Yevick hard
sphere pair distribution function, $g_2(r)$, for various spatial dimensions. We
find that beyond a certain critical volume fraction $\eta_c$, the pair
distribution function, $g_2(r)$, which should be positive definite, becomes
negative at some distances. We also present an intriguing observation that the
critical $\eta_c$ values we find are consistent with volume fractions where
onsets of random close packing (or maximally random jammed states) are reported
in the literature for various dimensions. That observation is supported by an
intuitive argument. This work may have important implications for other systems
for which a Percus-Yevick theory exists.
-
In the present work, crystallization in melts and poor-solvent solutions of
semiflexible polymers with different concentration was studied by means of
dissipative particle dynamics simulation technique. We use a coarse-grained
polymer model trying to catch general principles of crystallization in such
systems on large time and length scales. We observe the crystallization process
starting from an initial randomly prepared system with different polymer volume
fractions in a poor solvent. Because the solvent is very poor, the macrophase
polymer-solvent separation takes place very fast and is accompanied by partial
polymer crystallization. We have found that the overall crystalline fraction at
the end of crystallization process decreases upon increasing the polymer volume
fraction in the initial randomly prepared system, while the steady-state
crystallization speed is almost the same at polymer volume fractions larger
than 50\%. At the same time, the average crystallite size differs considerably
and has a maximum value in the systems with 90\% polymer volume fraction. We
assume that this polymer concentration is an optimal value in a sense of a
balance between the amount of polymer material available for increasing
crystallite size and chain entanglements preventing crystallites growth and
merging.
-
Equilibrium director structures in two thin hybrid planar films of biaxial
nematics are investigated through Markov chain Monte Carlo simulations based on
a lattice Hamiltonian model within the London dispersion approximation. While
the substrates of the two films induce similar anchoring influences on the long
axes of the liquid crystal molecules (viz. planar orientation at one end and
perpendicular, or homeotropic, orientations at the other), they differ in their
coupling with the minor axes of the molecules. In Type-A film the substrates do
not interact with the minor axes at all (which is experimentally relatively
more amenable), while in Type-B, the orientations of the molecular axes at the
surface layer are influenced as well by their biaxial coupling with the
surface. Both films exhibit expected bending of the director associated with
ordering of the molecular long axes due to surface anchoring. Simulation
results indicate that the Type-A film hosts stable and noise free director
structures in the biaxial nematic phase of the LC medium, resulting from
dominant ordering of one of the minor axes in the plane of the substrates. High
degree of this stable order thus developed could be of practical interest for
in-plane switching applications with an external field. Type-B film, on the
other hand, experiences competing interactions among the minor axes, due to
incompatible anchoring influences at the bounding substrates, apparently
leading to frustration, and hence to noisy equilibrium director structures.
-
This paper proposes a phase space to compare the static packings of a
granular system compatible to a macrostate that is set by the external stress.
The nature of this phase space is analyzed, showing that the consideration of
the allowed and forbidden regions and the internal degrees of freedom of every
configuration (i.e. geometrical pattern) could be a relevant factor for the
establishment of its probability and, therefore, of the expected properties of
the sample. This is due to the fact that many combinations of forces acting on
a particle can keep it in static equilibrium. Every set of forces can be
considered equivalent to a microscopic stress field, but the kind of
interaction and the geometrical restrictions mean that not all stress states
can be represented by any set, whereas others can be represented by many sets.
Consequently the points of the phase space are degenerate, and the density of
states of each configuration strongly determines the most probable statistical
distribution. It is shown how these functions just depend on the deviatoric
stress. A first analysis of two-dimensional (2D) arrangements is included to
clarify this assertion.
-
Spatially correlated noise (SCN), i.e. the thermal noise that affects
neighbouring particles in a similar manner, is ubiquitous in soft matter
systems. In this work, we apply the over-damped SCN-driven Langevin equations
as an effective, one-component model of the dynamics in dense binary mixtures.
We derive the thermodynamically consistent fluctuation-dissipation relation for
SCN to show that it predicts the molecular arrest resembling the glass
transition, i.e. the critical slow-down of dynamics in the disordered phases.
We show that the mechanism of singular dissipation is embedded in the
dissipation matrix, accompanying SCN. We are also able to identify the
characteristic length of collective dissipation, which diverges at critical
packing. This novel physical quantity conveniently describes the difference
between the ergodic and non-ergodic dynamics. The model is fully analytically
solvable, one-dimensional and admits arbitrary interactions between the
particles. It qualitatively reproduces several different modes of arrested
disorder encountered in binary mixtures, including e.g. the re-entrant arrest.
The model can be effectively compared to the mode coupling theory.
-
Many biological systems form colonies at high density. Passive granular
systems will be jammed at such densities, yet for the survival of biological
systems it is crucial that they are dynamic. We construct a phase diagram for a
system of active particles interacting via Vicsek alignment, and vary the
density, self-propulsion force, and orientational noise. We find that the
system exhibits four different phases, characterized by transitions in the
effective diffusion constant and in the orientational order parameter. Our
simulations show that there exists an optimal noise such that particles require
a minimal force to unjam, allowing for rearrangements.
-
The flow of colloidal suspensions is ubiquitous in nature and industry.
Colloidal suspensions exhibit a wide range of rheological behavior, which
should be closely related to the microscopic structure of the systems. With
in-situ small-angle neutron scattering complemented by rheological
measurements, we investigated the deformation behavior of a charge-stabilized
colloidal glass at particle level undergoing steady shear. A short-lived,
localized elastic response at particle level, termed as transient elasticity
zone (TEZ), was identified from the neutron spectra. The existence of the TEZ
is a signature of the dynamical heterogeneity: The body of fluids under shear
behaves like an elastic solid within the spatial range of TEZ but like fluid
outside the TEZ. The size of TEZ shrinks as the shear rate increases in the
shear thinning region, which shows that the shear thinning is accompanied by a
diminishing dynamical heterogeneity. More interestingly, the TEZ is found to be
the structural unit that provides the resistance to the imposed shear, as
evidenced by the quantitative agreement between the local elastic stress
sustained by TEZ and the macroscopic stress from rheological measurements at
low and moderate shear rates. Besides the charged-stabilized colloidal
suspension, a hard-sphere colloidal suspension at the same volume fraction and
shear rates was also measured. The result highlights the key role of the
electrostatic interparticle repulsion in promoting the local elasticity. Our
findings provide an understanding on the nonlinear rheology of interacting
colloidal glasses from a micro-mechanical view.
-
The formation of crystalline calcium sulfate (CaSO4*xH2O) polymorphs from
aqueous solutions is assumed to occur via a single-step process following the
classical nucleation paradigm. However, although recent research contradicts
this classical picture and indicates that CaSO4*2H2O forms at room temperature
through multiple steps at different length and time-scales, these steps have so
far not been quantified. By using in situ and fast time-resolved small angle
X-ray scattering (SAXS), we demonstrate that the nucleation and growth of
CaSO4*2H2O involves at the very initial stages the formation of well-defined,
primary species of < 3 nm in length (stage I). Stage II of the reaction is
characterized by the arrangement of these primary species into domains, while
in stage III these domains condense into larger aggregates. Based on volume
fractions and electron density considerations we propose that the fast forming
primary species from supersaturated aqueous CaSO4 solutions are composed of
anhydrous Ca-SO4-cores. The first three stages of nucleation and aggregation of
the primary species are followed by a final stage (stage IV), where the primary
species grow within the aggregates, and eventually transform into gypsum
(CaSO4*2H2O). This final stage was also confirmed through simultaneously
collected wide-angle scattering (diffraction, WAXS) data, which clearly show
the growth of gypsum during stage IV only. Our results demonstrate that CaSO4
formation is driven by the nucleation and aggregation of well-defined anhydrous
Ca-SO4-cores that transform through hydration into gypsum through a complex
nucleation and growth pathway.
-
Hypothesis: The peculiar swelling behaviour of poly(N-isopropylacrylamide)
(PNIPAM)-based responsive microgels provides the possibility to tune both
softness and volume fraction with temperature, making these systems of great
interest for technological applications and theoretical implications. Their
intriguing phase diagram can be even more complex if poly(acrylic acid) (PAAc)
is interpenetrated within PNIPAM network to form Interpenetrating Polymer
Network (IPN) microgels that exhibit an additional pH-sensitivity. The effect
of the PAAc/PNIPAM polymeric ratio on both swelling capability and dynamics is
still matter of investigation. Experiments: Here we investigate the role of
PAAc in the behaviour of IPN microgels across the volume phase transition
through dynamic light scattering (DLS), transmission electron microscopy (TEM)
and electrophoretic measurements as a function of microgel concentration and
pH. Findings: Our results highlight that aggregation is favored at increasing
weight concentration, PAAc content and pH and that a crossover PAAc content
C*_{PAAc} exists above which the ionic charges on the microgel become relevant.
Moreover we show that the softness of IPN microgels can be tuned ad hoc by
changing the PAAc/PNIPAM ratio. These findings provide new insights into the
possibility to control experimentally aggregation properties, charge and
softness of IPN microgels by varying PAAc content.
-
Cell division and death can be regulated by the mechanical forces within a
tissue. We study the consequences for the stability and roughness of a
propagating interface, by analysing a model of mechanically-regulated tissue
growth in the regime of small driving forces. For an interface driven by
homeostatic pressure imbalance or leader-cell motility, long and
intermediate-wavelength instabilities arise, depending respectively on an
effective viscosity of cell number change, and on substrate friction. A further
mechanism depends on the strength of directed motility forces acting in the
bulk. We analyse the fluctuations of a stable interface subjected to cell-level
stochasticity, and find that mechanical feedback can help preserve
reproducibility at the tissue scale. Our results elucidate mechanisms that
could be important for orderly interface motion in developing tissues.
-
The mesoscopic field theory for ionic systems [A. Ciach and G. Stell, J. Mol.
Liq. 87, 255 (2000)] is extended to the system with charged boundaries. A very
simple expression for the excess grand potential functional of the charge
density is developed. The size of hard-cores of ions is taken into account in
the expression for the internal energy. The functional is suitable for a
description of a distribution of ions in ionic liquids and ionic liquid
mixtures with neutral components near a weakly charged wall. The Euler-Lagrange
equation is obtained, and solved for a flat confining surface. An exponentially
damped oscillatory charge density profile is obtained. The electrostatic
potential for the restricted primitive model agrees with the simulation results
on a semiquantitative level.
-
Hydrodynamic interactions play an important role in many areas of soft matter
science. In simulations with implicit solvent, various techniques such as
Brownian or Stokesian dynamics explicitly include hydrodynamic interactions a
posteriori by using hydrodynamic diffusion tensors derived from the Stokes
equation. However, this equation assumes the interaction to be instantaneous
which is an idealized approximation and only valid on long time scales. In the
present paper, we go one step further and analyze the time-dependence of
hydrodynamic interactions in a compressible fluid on the basis of the
linearized Navier-Stokes equation. The theoretical results show that the
compressibility of the fluid has a significant impact on frequency-dependent
pair interactions.
The predictions of the hydrodynamic theory are compared to molecular dynamics
simulations of two solid spheres in a Lennard-Jones fluid. For this system we
reconstruct memory functions by extending the inverse Volterra technique. The
simulation data agree very well with the theory, therefore, the theory can be
used to implement dynamically consistent hydrodynamic interactions in the
increasingly popular field of non-Markovian modeling.
-
A self-replicator is usually understood to be an object of definite form that
promotes the conversion of materials in its environment into a nearly identical
copy of itself. The challenge of engineering novel, micro- or nano-scale
self-replicators has attracted keen interest in recent years, both because
exponential amplification is an attractive method for generating high yields of
specific products, and also because self-reproducing entities have the
potential to be optimized or adapted through rounds of iterative selection.
Substantial steps forward have been achieved both in the engineering of
particular self-replicating molecules, and also in characterizing the physical
basis for possible mechanisms of self-replication. At present, however, there
is need for a theoretical treatment of what physical conditions are most
conducive to the emergence of novel self-replicating structures from a
reservoir of building blocks on a desired time-scale. Here we report progress
in addressing this need. By analyzing the dynamics of a generic class of
heterogeneous particle mixtures whose reaction rates emerge from basic physical
interactions, we demonstrate that the spontaneous discovery of self-replication
is controlled by relatively generic features of the chemical space, namely: the
dispersion in the distribution of reaction timescales and bound-state energies.
Based on this analysis, we provide quantitative criteria that may aid
experimentalists in designing a system capable of producing self-replicators,
and in estimating the likely timescale for exponential growth to start.
-
Ultrasound is increasingly being used to modulate the properties of
biological membranes for applications in drug delivery and neuromodulation.
While various studies have investigated the mechanical aspect of the
interaction such as acoustic absorption and membrane deformation, it is not
clear how these effects transduce into biological functions, for example,
changes in the permeability or the enzymatic activity of the membrane. A
critical aspect of the activity of an enzyme is the thermal fluctuations of its
solvation or hydration shell. Thermal fluctuations are also known to be
directly related to membrane permeability. Here solvation shell changes of
lipid membranes subject to an acoustic impulse were investigated using a
fluorescence probe, Laurdan. Laurdan was embedded in multi-lamellar lipid
vesicles in water, which were exposed to broadband pressure impulses of the
order of 1MPa peak amplitude and 10{\mu}s pulse duration. An instrument was
developed to monitor changes in the emission spectrum of the dye at two
wavelengths with sub-microsecond temporal resolution. The experiments show that
changes in the emission spectrum, and hence the fluctuations of the solvation
shell, are related to the changes in the thermodynamic state of the membrane
and correlated with the compression and rarefaction of the incident sound wave.
The results suggest that acoustic fields affect the state of a lipid membrane
and therefore can potentially modulate the kinetics of channels and proteins
embedded in the membrane.
-
We investigate the geometrical and mechanical properties of adherent cells
characterized by a highly anisotropic actin cytoskeleton. Using a combination
of theoretical work and experiments on micropillar arrays, we demonstrate that
the shape of the cell edge is accurately described by elliptical arcs, whose
eccentricity expresses the degree of anisotropy of the internal cell stresses.
This results in a spatially varying tension along the cell edge, that
significantly affects the traction forces exerted by the cell on the substrate.
Our work highlights the strong interplay between cell mechanics and geometry
and paves the way towards the reconstruction of cellular forces from
geometrical data.
-
Active materials are media for which deformations can occur in absence of
loads, given an external stimulus. Two approaches to the modeling of such
materials are mainly used in literature, both based on the introduction of a
new tensor: an additive stress $\mathsf{P}_\text{act}$ in the active stress
case and a multiplicative strain $\mathsf{F}_a$ in the active strain one. Aim
of this paper is the comparison between the two approaches on simple shears.
Considering an incompressible and transversely isotropic material, we design
constitutive relations for $\mathsf{P}_\text{act}$ and $\mathsf{F}_a$ so that
they produce the same results for a uniaxial deformation along the symmetry
axis. We then study the two approaches in the case of a simple shear
deformation. In a hyperelastic setting, we show that the two approaches produce
different stress components along a simple shear, unless some necessary
conditions on the strain energy density are fulfilled. However, such conditions
are very restrictive and rule out the usual elastic strain energy functionals.
Active stress and active strain therefore produce different results in shear,
even if they both fit uniaxial data. Our results show that experimental data on
the stress-stretch response on uniaxial deformations are not enough to
establish which activation approach can capture better the mechanics of active
materials. We conclude that other types of deformations, beyond the uniaxial
one, should be taken into consideration in the modeling of such materials.
-
We consider the ferromagnetic Ising model on the Cayley tree and we
investigate the decomposition of the free state into extremal states below the
spin glass temperature. We show that this decomposition has uncountably many
components. The tail observable showing that the free state is not extremal is
related to the Edwards-Anderson parameter, measuring the variance of the
(random) magnetization obtained from drawing boundary conditions from the free
state.


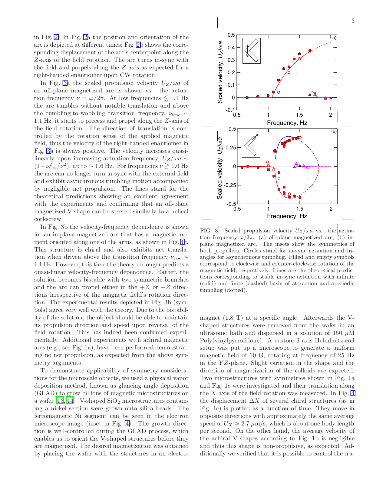
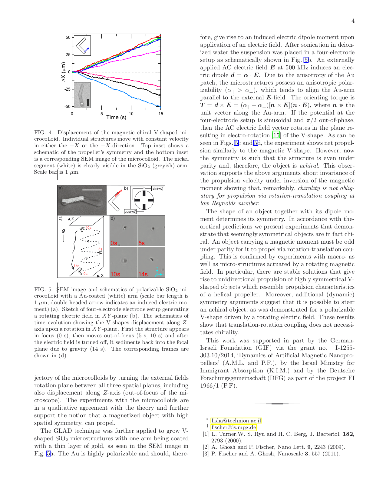

 We theoretically and experimentally investigate low-Reynolds-number propulsion of geometrically achiral planar objects that possess a dipole moment and that are driven by a rotating magnetic field. Symmetry considerations (involving parity, $\widehat{P}$, and charge conjugation, $\widehat{C}$) establish correspondence between propulsive states depending on orientation of the dipolar moment. Although basic symmetry arguments do not forbid individual symmetric objects to efficiently propel due to spontaneous symmetry breaking, they suggest that the average ensemble velocity vanishes. Some additional arguments show, however, that highly symmetrical ($\widehat{P}$-even) objects exhibit no net propulsion while individual less symmetrical ($\widehat{C}\widehat{P}$-even) propellers do propel. Particular magnetization orientation, rendering the shape $\widehat{C}\widehat{P}$-odd, yields unidirectional motion typically associated with chiral structures, such as helices. If instead of a structure with a permanent dipole we consider a polarizable object, some of the arguments have to be modified. For instance, we demonstrate a truly achiral ($\widehat{P}$- and $\widehat{C}\widehat{P}$-even) planar shape with an induced electric dipole that can propel by electro-rotation. We thereby show that chirality is not essential for propulsion due to rotation-translation coupling at low Reynolds number.
We theoretically and experimentally investigate low-Reynolds-number propulsion of geometrically achiral planar objects that possess a dipole moment and that are driven by a rotating magnetic field. Symmetry considerations (involving parity, $\widehat{P}$, and charge conjugation, $\widehat{C}$) establish correspondence between propulsive states depending on orientation of the dipolar moment. Although basic symmetry arguments do not forbid individual symmetric objects to efficiently propel due to spontaneous symmetry breaking, they suggest that the average ensemble velocity vanishes. Some additional arguments show, however, that highly symmetrical ($\widehat{P}$-even) objects exhibit no net propulsion while individual less symmetrical ($\widehat{C}\widehat{P}$-even) propellers do propel. Particular magnetization orientation, rendering the shape $\widehat{C}\widehat{P}$-odd, yields unidirectional motion typically associated with chiral structures, such as helices. If instead of a structure with a permanent dipole we consider a polarizable object, some of the arguments have to be modified. For instance, we demonstrate a truly achiral ($\widehat{P}$- and $\widehat{C}\widehat{P}$-even) planar shape with an induced electric dipole that can propel by electro-rotation. We thereby show that chirality is not essential for propulsion due to rotation-translation coupling at low Reynolds number.



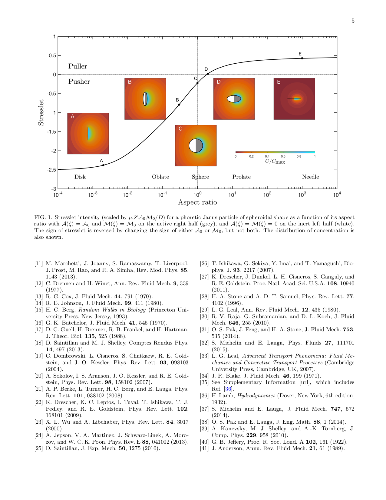
 Active particles disturb the fluid around them as force dipoles, or stresslets, which govern their collective dynamics. Unlike swimming speeds, the stresslets of active particles are rarely determined due to the lack of a suitable theoretical framework for arbitrary geometry. We propose a general method, based on the reciprocal theorem of Stokes flows, to compute stresslets as integrals of the velocities on the particle's surface, which we illustrate for spheroidal chemically-active particles. Our method will allow tuning the stresslet of artificial swimmers and tailoring their collective motion in complex environments.
Active particles disturb the fluid around them as force dipoles, or stresslets, which govern their collective dynamics. Unlike swimming speeds, the stresslets of active particles are rarely determined due to the lack of a suitable theoretical framework for arbitrary geometry. We propose a general method, based on the reciprocal theorem of Stokes flows, to compute stresslets as integrals of the velocities on the particle's surface, which we illustrate for spheroidal chemically-active particles. Our method will allow tuning the stresslet of artificial swimmers and tailoring their collective motion in complex environments.

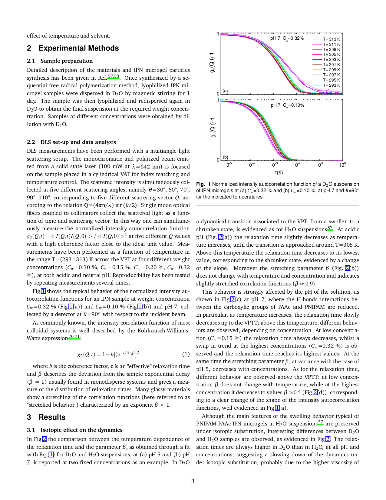
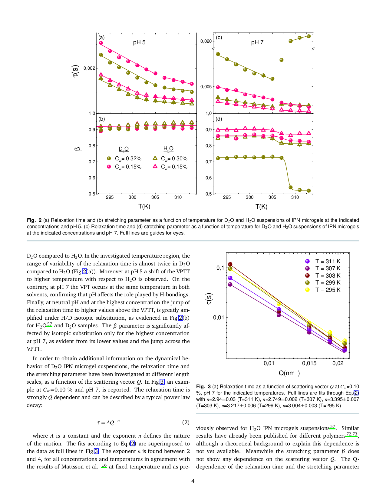
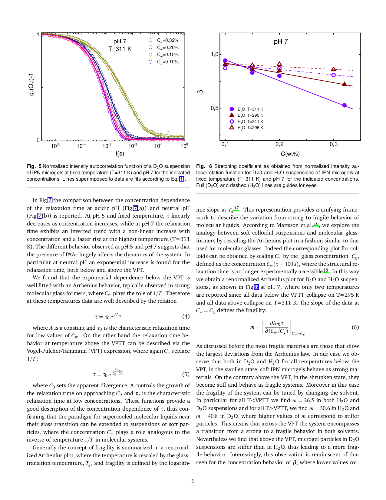 Microgel suspensions of Interpenetrated Polymer Network (IPN) of PNIPAM and PAAc in D$_2$O, have been investigated through dynamic light scattering as a function of temperature, pH and concentration across the Volume Phase Transition (VPT). The dynamics of the system is slowed down under H/D isotopic substitution due to the different balance between polymer/polymer and polymer/solvent interactions suggesting the crucial role played by H-bondings. The swelling behavior, reduced with respect to PNIPAM and water, has been described by the Flory-Rehner theory, tested for PNIPAM microgel and successfully expanded to higher order for IPN microgels. Moreover the concentration dependence of the relaxation time at neutral pH has highlighted two different routes to approach the glass transition: Arrhenius and super-Arrhenius (Vogel Fulcher Tammann) respectively below and above the VPT and a fragility plot has been derived. Fragility can be tuned by changing temperature: across the VPT particles undergo a transition from soft-strong to stiff-fragile.
Microgel suspensions of Interpenetrated Polymer Network (IPN) of PNIPAM and PAAc in D$_2$O, have been investigated through dynamic light scattering as a function of temperature, pH and concentration across the Volume Phase Transition (VPT). The dynamics of the system is slowed down under H/D isotopic substitution due to the different balance between polymer/polymer and polymer/solvent interactions suggesting the crucial role played by H-bondings. The swelling behavior, reduced with respect to PNIPAM and water, has been described by the Flory-Rehner theory, tested for PNIPAM microgel and successfully expanded to higher order for IPN microgels. Moreover the concentration dependence of the relaxation time at neutral pH has highlighted two different routes to approach the glass transition: Arrhenius and super-Arrhenius (Vogel Fulcher Tammann) respectively below and above the VPT and a fragility plot has been derived. Fragility can be tuned by changing temperature: across the VPT particles undergo a transition from soft-strong to stiff-fragile.



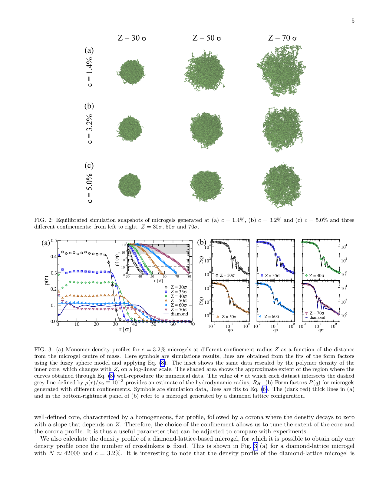
 Microgels are colloidal-scale particles individually made of crosslinked polymer networks that can swell and deswell in response to external stimuli, such as changes to temperature or pH. Despite a large amount of experimental activities on microgels, a proper theoretical description based on individual particle properties is still missing due to the complexity of the particles. To go one step further, here we propose a novel methodology to assemble realistic microgel particles "in silico". We exploit the self-assembly of a binary mixture composed of tetravalent (crosslinkers) and bivalent (monomer beads) patchy particles under spherical confinement in order to produce fully-bonded networks. The resulting structure is then used to generate the initial microgel configuration, which is subsequently simulated with a bead-spring model complemented by a temperature-induced hydrophobic attraction. To validate our assembly protocol we focus on a small microgel test-case and show that we can reproduce the experimental swelling curve by appropriately tuning the confining sphere radius, something that would not be possible with less sophisticated assembly methodologies, e.g. in the case of networks generated from an underlying crystal structure. We further investigate the structure (in reciprocal and real space) and the swelling curves of microgels as a function of temperature, finding that our results are well described by the widely-used fuzzy sphere model. This is a first step toward a realistic modelling of microgel particles, which will pave the way for a careful assessment of their elastic properties and effective interactions.
Microgels are colloidal-scale particles individually made of crosslinked polymer networks that can swell and deswell in response to external stimuli, such as changes to temperature or pH. Despite a large amount of experimental activities on microgels, a proper theoretical description based on individual particle properties is still missing due to the complexity of the particles. To go one step further, here we propose a novel methodology to assemble realistic microgel particles "in silico". We exploit the self-assembly of a binary mixture composed of tetravalent (crosslinkers) and bivalent (monomer beads) patchy particles under spherical confinement in order to produce fully-bonded networks. The resulting structure is then used to generate the initial microgel configuration, which is subsequently simulated with a bead-spring model complemented by a temperature-induced hydrophobic attraction. To validate our assembly protocol we focus on a small microgel test-case and show that we can reproduce the experimental swelling curve by appropriately tuning the confining sphere radius, something that would not be possible with less sophisticated assembly methodologies, e.g. in the case of networks generated from an underlying crystal structure. We further investigate the structure (in reciprocal and real space) and the swelling curves of microgels as a function of temperature, finding that our results are well described by the widely-used fuzzy sphere model. This is a first step toward a realistic modelling of microgel particles, which will pave the way for a careful assessment of their elastic properties and effective interactions.
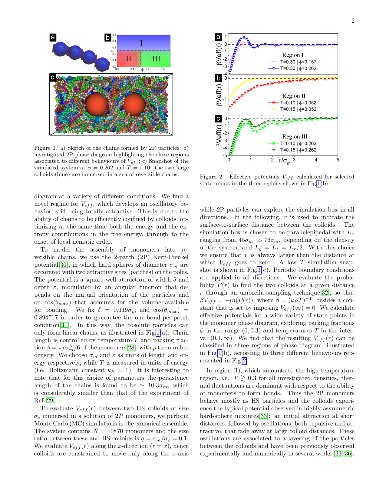



 Effective colloid-colloid interactions can be tailored through the addition of a complex cosolute. Here we investigate the case of a cosolute made by self-assembling patchy particles. Depending on the valence, these particles can form either polymer chains or branched structures. We numerically calculate the effective potential $V_{eff}$ between two colloids immersed in a suspension of reversible patchy particles, exploring a wide region of the cosolute phase diagram and the role of valence. In addition to well-known excluded volume and depletion effects, we find that, under appropriate conditions, $V_{eff}$ is completely attractive but shows an oscillatory character. In the case of polymerizing cosolute, this results from the fact that chains are efficiently confined by the colloids through the onset of local order. This argument is then generalized to the case of particles with higher valence, under the condition that they are still able to maintain a fully bonded organization upon confinement. The resulting effective potentials are relevant for understanding the behavior of complex mixtures in crowded environments, but may also be exploited for tuning colloidal self-assembly at preferred target distances in order to build desired superstructures.
Effective colloid-colloid interactions can be tailored through the addition of a complex cosolute. Here we investigate the case of a cosolute made by self-assembling patchy particles. Depending on the valence, these particles can form either polymer chains or branched structures. We numerically calculate the effective potential $V_{eff}$ between two colloids immersed in a suspension of reversible patchy particles, exploring a wide region of the cosolute phase diagram and the role of valence. In addition to well-known excluded volume and depletion effects, we find that, under appropriate conditions, $V_{eff}$ is completely attractive but shows an oscillatory character. In the case of polymerizing cosolute, this results from the fact that chains are efficiently confined by the colloids through the onset of local order. This argument is then generalized to the case of particles with higher valence, under the condition that they are still able to maintain a fully bonded organization upon confinement. The resulting effective potentials are relevant for understanding the behavior of complex mixtures in crowded environments, but may also be exploited for tuning colloidal self-assembly at preferred target distances in order to build desired superstructures.




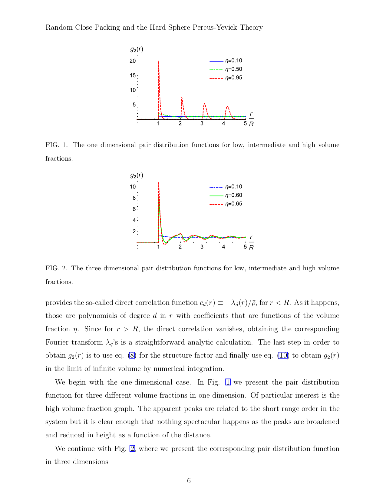 The Percus-Yevick theory for monodisperse hard spheres gives very good results for the pressure and structure factor of the system in a whole range of densities that lie within the liquid phase. However, the equation seems to lead to a very unacceptable result beyond that region. Namely, the Percus-Yevick theory predicts a smooth behavior of the pressure that diverges only when the volume fraction $\eta$ approaches unity. Thus, within the theory there seems to be no indication for the termination of the liquid phase and the transition to a solid or to a glass. In the present article we study the Percus-Yevick hard sphere pair distribution function, $g_2(r)$, for various spatial dimensions. We find that beyond a certain critical volume fraction $\eta_c$, the pair distribution function, $g_2(r)$, which should be positive definite, becomes negative at some distances. We also present an intriguing observation that the critical $\eta_c$ values we find are consistent with volume fractions where onsets of random close packing (or maximally random jammed states) are reported in the literature for various dimensions. That observation is supported by an intuitive argument. This work may have important implications for other systems for which a Percus-Yevick theory exists.
The Percus-Yevick theory for monodisperse hard spheres gives very good results for the pressure and structure factor of the system in a whole range of densities that lie within the liquid phase. However, the equation seems to lead to a very unacceptable result beyond that region. Namely, the Percus-Yevick theory predicts a smooth behavior of the pressure that diverges only when the volume fraction $\eta$ approaches unity. Thus, within the theory there seems to be no indication for the termination of the liquid phase and the transition to a solid or to a glass. In the present article we study the Percus-Yevick hard sphere pair distribution function, $g_2(r)$, for various spatial dimensions. We find that beyond a certain critical volume fraction $\eta_c$, the pair distribution function, $g_2(r)$, which should be positive definite, becomes negative at some distances. We also present an intriguing observation that the critical $\eta_c$ values we find are consistent with volume fractions where onsets of random close packing (or maximally random jammed states) are reported in the literature for various dimensions. That observation is supported by an intuitive argument. This work may have important implications for other systems for which a Percus-Yevick theory exists.




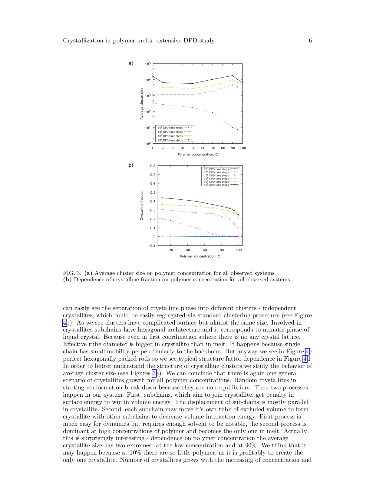 In the present work, crystallization in melts and poor-solvent solutions of semiflexible polymers with different concentration was studied by means of dissipative particle dynamics simulation technique. We use a coarse-grained polymer model trying to catch general principles of crystallization in such systems on large time and length scales. We observe the crystallization process starting from an initial randomly prepared system with different polymer volume fractions in a poor solvent. Because the solvent is very poor, the macrophase polymer-solvent separation takes place very fast and is accompanied by partial polymer crystallization. We have found that the overall crystalline fraction at the end of crystallization process decreases upon increasing the polymer volume fraction in the initial randomly prepared system, while the steady-state crystallization speed is almost the same at polymer volume fractions larger than 50\%. At the same time, the average crystallite size differs considerably and has a maximum value in the systems with 90\% polymer volume fraction. We assume that this polymer concentration is an optimal value in a sense of a balance between the amount of polymer material available for increasing crystallite size and chain entanglements preventing crystallites growth and merging.
In the present work, crystallization in melts and poor-solvent solutions of semiflexible polymers with different concentration was studied by means of dissipative particle dynamics simulation technique. We use a coarse-grained polymer model trying to catch general principles of crystallization in such systems on large time and length scales. We observe the crystallization process starting from an initial randomly prepared system with different polymer volume fractions in a poor solvent. Because the solvent is very poor, the macrophase polymer-solvent separation takes place very fast and is accompanied by partial polymer crystallization. We have found that the overall crystalline fraction at the end of crystallization process decreases upon increasing the polymer volume fraction in the initial randomly prepared system, while the steady-state crystallization speed is almost the same at polymer volume fractions larger than 50\%. At the same time, the average crystallite size differs considerably and has a maximum value in the systems with 90\% polymer volume fraction. We assume that this polymer concentration is an optimal value in a sense of a balance between the amount of polymer material available for increasing crystallite size and chain entanglements preventing crystallites growth and merging.




 Equilibrium director structures in two thin hybrid planar films of biaxial nematics are investigated through Markov chain Monte Carlo simulations based on a lattice Hamiltonian model within the London dispersion approximation. While the substrates of the two films induce similar anchoring influences on the long axes of the liquid crystal molecules (viz. planar orientation at one end and perpendicular, or homeotropic, orientations at the other), they differ in their coupling with the minor axes of the molecules. In Type-A film the substrates do not interact with the minor axes at all (which is experimentally relatively more amenable), while in Type-B, the orientations of the molecular axes at the surface layer are influenced as well by their biaxial coupling with the surface. Both films exhibit expected bending of the director associated with ordering of the molecular long axes due to surface anchoring. Simulation results indicate that the Type-A film hosts stable and noise free director structures in the biaxial nematic phase of the LC medium, resulting from dominant ordering of one of the minor axes in the plane of the substrates. High degree of this stable order thus developed could be of practical interest for in-plane switching applications with an external field. Type-B film, on the other hand, experiences competing interactions among the minor axes, due to incompatible anchoring influences at the bounding substrates, apparently leading to frustration, and hence to noisy equilibrium director structures.
Equilibrium director structures in two thin hybrid planar films of biaxial nematics are investigated through Markov chain Monte Carlo simulations based on a lattice Hamiltonian model within the London dispersion approximation. While the substrates of the two films induce similar anchoring influences on the long axes of the liquid crystal molecules (viz. planar orientation at one end and perpendicular, or homeotropic, orientations at the other), they differ in their coupling with the minor axes of the molecules. In Type-A film the substrates do not interact with the minor axes at all (which is experimentally relatively more amenable), while in Type-B, the orientations of the molecular axes at the surface layer are influenced as well by their biaxial coupling with the surface. Both films exhibit expected bending of the director associated with ordering of the molecular long axes due to surface anchoring. Simulation results indicate that the Type-A film hosts stable and noise free director structures in the biaxial nematic phase of the LC medium, resulting from dominant ordering of one of the minor axes in the plane of the substrates. High degree of this stable order thus developed could be of practical interest for in-plane switching applications with an external field. Type-B film, on the other hand, experiences competing interactions among the minor axes, due to incompatible anchoring influences at the bounding substrates, apparently leading to frustration, and hence to noisy equilibrium director structures.




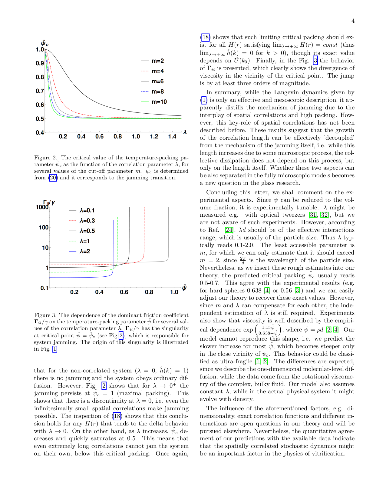
 This paper proposes a phase space to compare the static packings of a granular system compatible to a macrostate that is set by the external stress. The nature of this phase space is analyzed, showing that the consideration of the allowed and forbidden regions and the internal degrees of freedom of every configuration (i.e. geometrical pattern) could be a relevant factor for the establishment of its probability and, therefore, of the expected properties of the sample. This is due to the fact that many combinations of forces acting on a particle can keep it in static equilibrium. Every set of forces can be considered equivalent to a microscopic stress field, but the kind of interaction and the geometrical restrictions mean that not all stress states can be represented by any set, whereas others can be represented by many sets. Consequently the points of the phase space are degenerate, and the density of states of each configuration strongly determines the most probable statistical distribution. It is shown how these functions just depend on the deviatoric stress. A first analysis of two-dimensional (2D) arrangements is included to clarify this assertion.
This paper proposes a phase space to compare the static packings of a granular system compatible to a macrostate that is set by the external stress. The nature of this phase space is analyzed, showing that the consideration of the allowed and forbidden regions and the internal degrees of freedom of every configuration (i.e. geometrical pattern) could be a relevant factor for the establishment of its probability and, therefore, of the expected properties of the sample. This is due to the fact that many combinations of forces acting on a particle can keep it in static equilibrium. Every set of forces can be considered equivalent to a microscopic stress field, but the kind of interaction and the geometrical restrictions mean that not all stress states can be represented by any set, whereas others can be represented by many sets. Consequently the points of the phase space are degenerate, and the density of states of each configuration strongly determines the most probable statistical distribution. It is shown how these functions just depend on the deviatoric stress. A first analysis of two-dimensional (2D) arrangements is included to clarify this assertion.

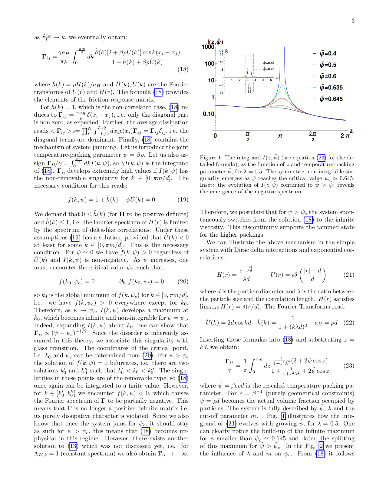

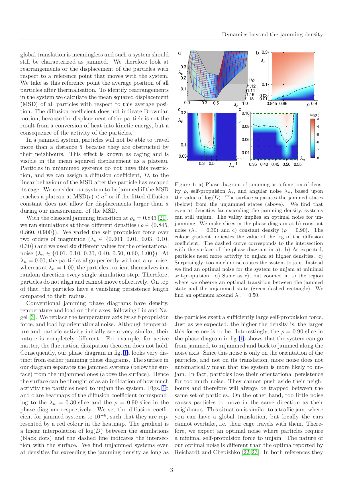
 Spatially correlated noise (SCN), i.e. the thermal noise that affects neighbouring particles in a similar manner, is ubiquitous in soft matter systems. In this work, we apply the over-damped SCN-driven Langevin equations as an effective, one-component model of the dynamics in dense binary mixtures. We derive the thermodynamically consistent fluctuation-dissipation relation for SCN to show that it predicts the molecular arrest resembling the glass transition, i.e. the critical slow-down of dynamics in the disordered phases. We show that the mechanism of singular dissipation is embedded in the dissipation matrix, accompanying SCN. We are also able to identify the characteristic length of collective dissipation, which diverges at critical packing. This novel physical quantity conveniently describes the difference between the ergodic and non-ergodic dynamics. The model is fully analytically solvable, one-dimensional and admits arbitrary interactions between the particles. It qualitatively reproduces several different modes of arrested disorder encountered in binary mixtures, including e.g. the re-entrant arrest. The model can be effectively compared to the mode coupling theory.
Spatially correlated noise (SCN), i.e. the thermal noise that affects neighbouring particles in a similar manner, is ubiquitous in soft matter systems. In this work, we apply the over-damped SCN-driven Langevin equations as an effective, one-component model of the dynamics in dense binary mixtures. We derive the thermodynamically consistent fluctuation-dissipation relation for SCN to show that it predicts the molecular arrest resembling the glass transition, i.e. the critical slow-down of dynamics in the disordered phases. We show that the mechanism of singular dissipation is embedded in the dissipation matrix, accompanying SCN. We are also able to identify the characteristic length of collective dissipation, which diverges at critical packing. This novel physical quantity conveniently describes the difference between the ergodic and non-ergodic dynamics. The model is fully analytically solvable, one-dimensional and admits arbitrary interactions between the particles. It qualitatively reproduces several different modes of arrested disorder encountered in binary mixtures, including e.g. the re-entrant arrest. The model can be effectively compared to the mode coupling theory.

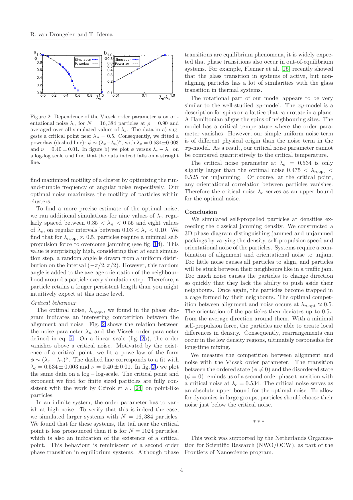

 Many biological systems form colonies at high density. Passive granular systems will be jammed at such densities, yet for the survival of biological systems it is crucial that they are dynamic. We construct a phase diagram for a system of active particles interacting via Vicsek alignment, and vary the density, self-propulsion force, and orientational noise. We find that the system exhibits four different phases, characterized by transitions in the effective diffusion constant and in the orientational order parameter. Our simulations show that there exists an optimal noise such that particles require a minimal force to unjam, allowing for rearrangements.
Many biological systems form colonies at high density. Passive granular systems will be jammed at such densities, yet for the survival of biological systems it is crucial that they are dynamic. We construct a phase diagram for a system of active particles interacting via Vicsek alignment, and vary the density, self-propulsion force, and orientational noise. We find that the system exhibits four different phases, characterized by transitions in the effective diffusion constant and in the orientational order parameter. Our simulations show that there exists an optimal noise such that particles require a minimal force to unjam, allowing for rearrangements.

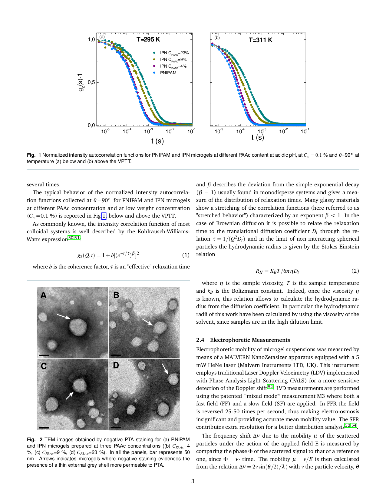
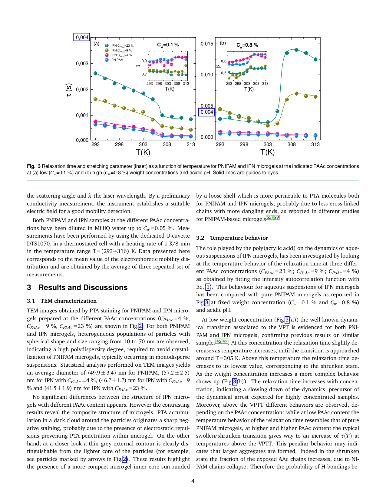
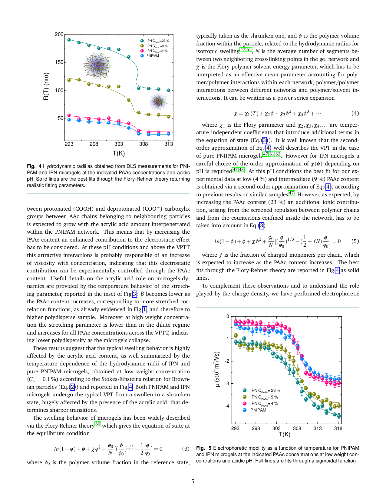
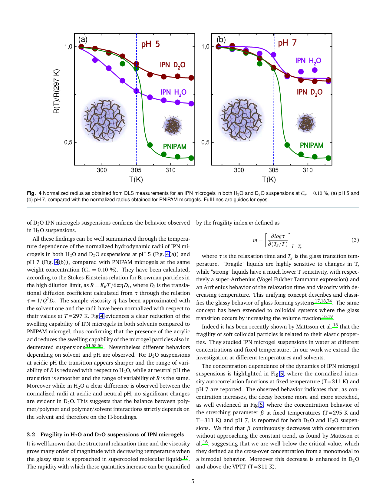
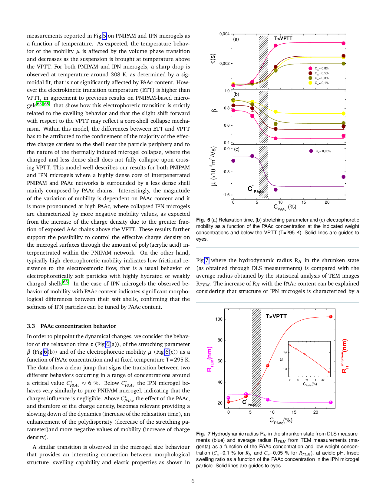 Hypothesis: The peculiar swelling behaviour of poly(N-isopropylacrylamide) (PNIPAM)-based responsive microgels provides the possibility to tune both softness and volume fraction with temperature, making these systems of great interest for technological applications and theoretical implications. Their intriguing phase diagram can be even more complex if poly(acrylic acid) (PAAc) is interpenetrated within PNIPAM network to form Interpenetrating Polymer Network (IPN) microgels that exhibit an additional pH-sensitivity. The effect of the PAAc/PNIPAM polymeric ratio on both swelling capability and dynamics is still matter of investigation. Experiments: Here we investigate the role of PAAc in the behaviour of IPN microgels across the volume phase transition through dynamic light scattering (DLS), transmission electron microscopy (TEM) and electrophoretic measurements as a function of microgel concentration and pH. Findings: Our results highlight that aggregation is favored at increasing weight concentration, PAAc content and pH and that a crossover PAAc content C*_{PAAc} exists above which the ionic charges on the microgel become relevant. Moreover we show that the softness of IPN microgels can be tuned ad hoc by changing the PAAc/PNIPAM ratio. These findings provide new insights into the possibility to control experimentally aggregation properties, charge and softness of IPN microgels by varying PAAc content.
Hypothesis: The peculiar swelling behaviour of poly(N-isopropylacrylamide) (PNIPAM)-based responsive microgels provides the possibility to tune both softness and volume fraction with temperature, making these systems of great interest for technological applications and theoretical implications. Their intriguing phase diagram can be even more complex if poly(acrylic acid) (PAAc) is interpenetrated within PNIPAM network to form Interpenetrating Polymer Network (IPN) microgels that exhibit an additional pH-sensitivity. The effect of the PAAc/PNIPAM polymeric ratio on both swelling capability and dynamics is still matter of investigation. Experiments: Here we investigate the role of PAAc in the behaviour of IPN microgels across the volume phase transition through dynamic light scattering (DLS), transmission electron microscopy (TEM) and electrophoretic measurements as a function of microgel concentration and pH. Findings: Our results highlight that aggregation is favored at increasing weight concentration, PAAc content and pH and that a crossover PAAc content C*_{PAAc} exists above which the ionic charges on the microgel become relevant. Moreover we show that the softness of IPN microgels can be tuned ad hoc by changing the PAAc/PNIPAM ratio. These findings provide new insights into the possibility to control experimentally aggregation properties, charge and softness of IPN microgels by varying PAAc content.

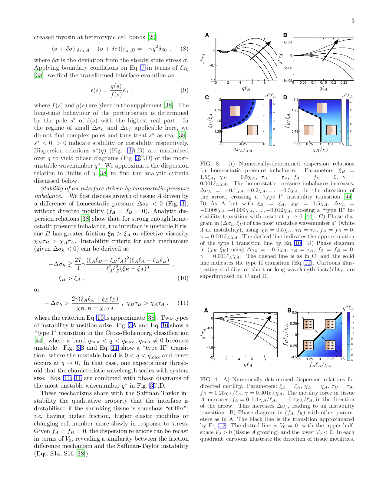


 Cell division and death can be regulated by the mechanical forces within a tissue. We study the consequences for the stability and roughness of a propagating interface, by analysing a model of mechanically-regulated tissue growth in the regime of small driving forces. For an interface driven by homeostatic pressure imbalance or leader-cell motility, long and intermediate-wavelength instabilities arise, depending respectively on an effective viscosity of cell number change, and on substrate friction. A further mechanism depends on the strength of directed motility forces acting in the bulk. We analyse the fluctuations of a stable interface subjected to cell-level stochasticity, and find that mechanical feedback can help preserve reproducibility at the tissue scale. Our results elucidate mechanisms that could be important for orderly interface motion in developing tissues.
Cell division and death can be regulated by the mechanical forces within a tissue. We study the consequences for the stability and roughness of a propagating interface, by analysing a model of mechanically-regulated tissue growth in the regime of small driving forces. For an interface driven by homeostatic pressure imbalance or leader-cell motility, long and intermediate-wavelength instabilities arise, depending respectively on an effective viscosity of cell number change, and on substrate friction. A further mechanism depends on the strength of directed motility forces acting in the bulk. We analyse the fluctuations of a stable interface subjected to cell-level stochasticity, and find that mechanical feedback can help preserve reproducibility at the tissue scale. Our results elucidate mechanisms that could be important for orderly interface motion in developing tissues.
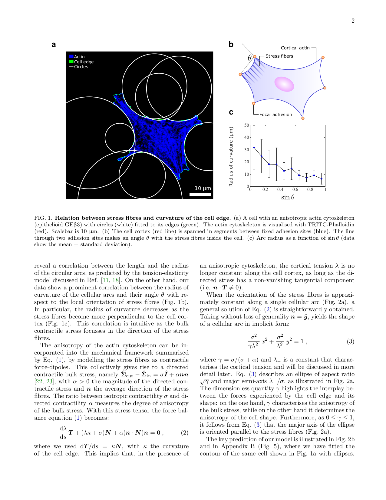
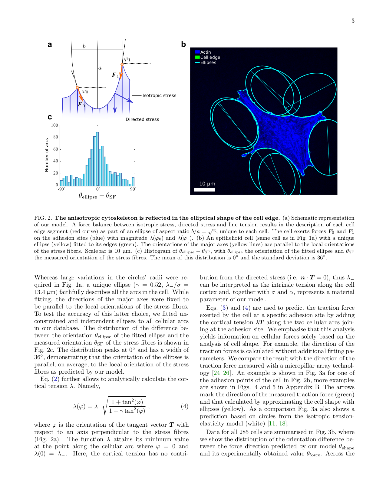
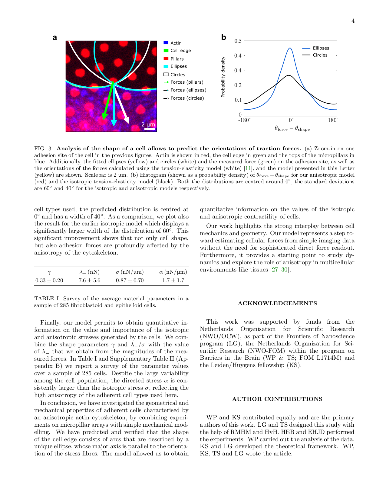

 We investigate the geometrical and mechanical properties of adherent cells characterized by a highly anisotropic actin cytoskeleton. Using a combination of theoretical work and experiments on micropillar arrays, we demonstrate that the shape of the cell edge is accurately described by elliptical arcs, whose eccentricity expresses the degree of anisotropy of the internal cell stresses. This results in a spatially varying tension along the cell edge, that significantly affects the traction forces exerted by the cell on the substrate. Our work highlights the strong interplay between cell mechanics and geometry and paves the way towards the reconstruction of cellular forces from geometrical data.
We investigate the geometrical and mechanical properties of adherent cells characterized by a highly anisotropic actin cytoskeleton. Using a combination of theoretical work and experiments on micropillar arrays, we demonstrate that the shape of the cell edge is accurately described by elliptical arcs, whose eccentricity expresses the degree of anisotropy of the internal cell stresses. This results in a spatially varying tension along the cell edge, that significantly affects the traction forces exerted by the cell on the substrate. Our work highlights the strong interplay between cell mechanics and geometry and paves the way towards the reconstruction of cellular forces from geometrical data.