-
Photoisomerization in a system with multiple electronic states and anharmonic
potential surfaces in a dissipative environment is investigated using a
rigorous numerical method employing quantum hierarchical Fokker-Planck
equations (QHFPE) for multi-state systems. We have developed a computer code
incorporating QHFPE for general-purpose computing on graphics processing units
(GPGPU) that can treat multi-state systems in phase space with any strength of
diabatic coupling of electronic states under non-perturbative and non-Markovian
system-bath interactions. This approach facilitates the calculation of both
linear and nonlinear spectra. We computed Wigner distributions for excited,
ground, and coherent states. We then investigated excited state dynamics
involving transitions among these states by analyzing linear absorption and
transient absorption processes and multi-dimensional electronic spectra with
various values of the heat bath parameters. Our results provide predictions for
spectroscopic measurements of photoisomerization dynamics. The motion of
excitation and ground state wavepackets and their coherence involved in the
photoisomerization were observed as the profiles of positive and negative peaks
of two-dimensional spectra.
-
Methyl cyanide is an important trace molecule in space, especially in
star-forming regions where it is one of the more common molecules used to
derive kinetic temperatures. We want to obtain accurate spectroscopic
parameters of minor isotopologs of methyl cyanide in their lowest excited $v_8
= 1$ vibrational states to support astronomical observations, in particular,
with interferometers such as ALMA. The laboratory rotational spectrum of methyl
cyanide in natural isotopic composition has been recorded from the millimeter
to the terahertz regions. Transitions with good signal-to-noise ratios could be
identified for the three isotopic species CH$_3^{13}$CN, $^{13}$CH$_3$CN, and
CH$_3$C(15)N up to about 1.2 THz ($J'' \le 66$). Accurate spectroscopic
parameters were obtained for all three species. The present data were already
instrumental in identifying $v_8 = 1$ lines of methyl cyanide with one $^{13}$C
in IRAM 30 m and ALMA data toward Sagittarius B2(N).
-
Ligand diffusion through proteins is a fundamental process governing
biological signaling and enzymatic catalysis. The complex topology of protein
tunnels results in difficulties with computing ligand escape pathways by
standard molecular dynamics (MD) simulations. Here, two novel methods for
searching of ligand exit pathways and cavity exploration are proposed: memory
random acceleration MD (mRAMD), and memetic algorithms (MA). In mRAMD, finding
exit pathways is based on a non-Markovian biasing that is introduced to
optimize the unbinding force. In MA, hybrid learning protocols are exploited to
predict optimal ligand exit paths. The methods are tested on three proteins
with increasing complexity of tunnels: M2 muscarinic receptor, nitrile
hydratase, and cytochrome P450cam. In these cases, the proposed methods
outperform standard techniques that are used currently to find ligand egress
pathways. The proposed approach is general and appropriate for accelerated
transport of an object through a network of protein tunnels.
-
Rotational transitions of $iso$-propyl cyanide, (CH$_3$)$_2$CHCN, also known
as $iso$-butyronitrile, were recorded using long-path absorption spectroscopy
in selected regions between 37 and 600 GHz. Further measurements were carried
out between 6 and 20 GHz employing Fourier transform microwave (FTMW)
spectroscopy on a pulsed molecular supersonic jet. The observed transitions
reach $J$ and $K_a$ quantum numbers of 103 and 59, respectively, and yield
accurate rotational constants as well as distortion parameters up to eighth
order. The $^{14}$N nuclear hyperfine splitting was resolved in particular by
FTMW spectroscopy yielding spin-rotation parameters as well as very accurate
quadrupole coupling terms. In addition, Stark effect measurements were carried
out in the microwave region to obtain a largely revised $c$-dipole moment
component and to improve the $a$-component. The hyperfine coupling and dipole
moment values are compared with values for related molecules both from
experiment and from quantum chemical calculations.
-
Methyl cyanide is an important trace molecule in star-forming regions. It is
one of the more common molecules used to derive kinetic temperatures in such
sources. As preparatory work for Herschel, SOFIA, and in particular ALMA we
want to improve the rest frequencies of the main as well as minor isotopologs
of methyl cyanide. The laboratory rotational spectrum of methyl cyanide in
natural isotopic composition has been recorded up to 1.63 THz. Transitions with
good signal-to-noise ratio could be identified for CH$_3$CN, $^{13}$CH$_3$CN,
CH$_3^{13}$CN, CH$_3$C$^{15}$N, CH$_2$DCN, and $^{13}$CH$_3^{13}$CN in their
ground vibrational states up to about 1.2 THz. The main isotopic species could
be identified even in the highest frequency spectral recordings around 1.6 THz.
The highest $J'$ quantum numbers included in the fit are 64 for
$^{13}$CH$_3^{13}$CN and 89 for the main isotopic species. Greatly improved
spectroscopic parameters have been obtained by fitting the present data
together with previously reported transition frequencies. The present data will
be helpful to identify isotopologs of methyl cyanide in the higher frequency
bands of instruments such as the recently launched Herschel satellite, the
upcoming airplane mission SOFIA or the radio telescope array ALMA.
-
The rotational spectra of the two stable conformers syn- and gauche-propanal
(CH$_3$CH$_2$CHO) were studied in the millimeter and submillimeter wave regions
from 75 to 500 GHz with the Cologne (Sub-)Millimeter wave Spectrometer.
Furthermore, the first excited states associated with the aldehyde torsion and
with the methyl torsion, respectively, of the $syn$-conformer were analyzed.
The newly obtained spectroscopic parameters yield better predictions, thus
fulfill sensitivity and resolution requirements in new astronomical
observations in order to unambiguously assign pure rotational transitions of
propanal. This is demonstrated on a radio astronomical spectrum from the
Atacama Large Millimeter/submillimeter Array Protostellar Interferometric Line
Survey (ALMA-PILS). In particular, an accurate description of observed
splittings, caused by internal rotation of the methyl group in the
$syn$-conformer and by tunneling rotation interaction from two stable
degenerate $gauche$-conformers, is reported. The rotational spectrum of
propanal is of additional interest because of its two large amplitude motions
pertaining to the methyl and the aldehyde group, respectively.
-
Spectra of methyl cyanide were recorded to analyze interactions in low-lying
vibrational states and to construct line lists for radio astronomical
observations as well as for infrared spectroscopic investigations of planetary
atmospheres. The rotational spectra cover large portions of the 36$-$1627 GHz
region. In the infrared (IR), a spectrum was recorded for this study in the
region of 2$\nu _8$ around 717 cm$^{-1}$ with assignments covering 684$-$765
cm$^{-1}$. Additional spectra in the $\nu _8$ region were used to validate the
analysis.
The large amount and the high accuracy of the rotational data extend to much
higher $J$ and $K$ quantum numbers and allowed us to investigate for the first
time in depth local interactions between these states which occur at high $K$
values. In particular, we have detected several interactions between $v_8 = 1$
and 2. Notably, there is a strong $\Delta v_8 = \pm1$, $\Delta K = 0$, $\Delta
l = \pm3$ Fermi resonance between $v_8 = 1^{-1}$ and $v_8 = 2^{+2}$ at $K$ =
14. Pronounced effects in the spectrum are also caused by resonant $\Delta v_8
= \pm1$, $\Delta K = \mp2$, $\Delta l = \pm1$ interactions between $v_8 = 1$
and 2. An equivalent resonant interaction occurs between $K$ = 14 of the ground
vibrational state and $K$ = 12, $l = +1$ of $v_8 = 1$ for which we present the
first detailed account. A preliminary account was given in an earlier study on
the ground vibrational state. From data pertaining to $v_8 = 2$, we also
investigated rotational interactions with $v_4 = 1$ as well as $\Delta v_8 =
\pm1$, $\Delta K = 0$, $\Delta l = \pm3$ Fermi interactions between $v_8 = 2$
and 3.
We have derived N$_2$- and self-broadening coefficients for the $\nu _8$,
2$\nu _8 - \nu _8$, and 2$\nu _8$ bands from previously determined nu4 values.
Subsequently, we determined transition moments and intensities for the three IR
bands.
-
The rotational spectrum of the formaldehyde isotopologue H$_2$C$^{17}$O was
investigated between 0.56 and 1.50 THz using a sample of natural isotopic
composition. In addition, transition frequencies were determined for
H$_2$C$^{18}$O and H$_2$C$^{16}$O between 1.37 and 1.50 THz. The data were
combined with critically evaluated literature data to derive improved sets of
spectroscopic parameters which include $^{17}$O or H nuclear hyperfine
structure parameters.
-
Density functional theory (DFT) calculation has had huge success as a tool
capable of predicting important physical and chemical properties of condensed
matter systems. We calculate the electric dipole moment of a molecule by using
the differential electron density with respect to the superimposed electron
density of the free atoms, exploiting the cancellation of DFT errors. Our
results on a range of molecules show an excellent agreement with experiments.
-
We present a new state function, denotes the energy of thermal motion, it
shows that the energy classification for the internal energy has been
completed. In a new theoretical structure, we present the concept of the heat
partial pressure, rewrite the equations of the first law, by which, the energy
conversion and energy transport can be distinguished by an explicit way, we
redefine the concept of the entropy, and confirm a conversion potential related
to the dissipative compensation, we discuss in detail on the three sources of
irreversibility, and the conversions between the calorimetric entropy and the
configurational entropy, an interesting conclusion shows that the second law
itself has contained the mechanism of evolution.
-
Engineered $Na_{0.75}CoO_2$ is considered a prime candidate to achieve
high-efficiency thermoelectric systems to regenerate electricity from waste
heat. In this work, three elements with outmost electronic configurations, (1)
an open d shell (Ni), (2) a closed d shell (Zn), and (3) a half filled f shell
(Eu) with maximum unpaired electrons, were selected to outline the dopants'
effects on electronic and crystallographic structures of $Na_{0.75}CoO_2$.
Systematic $ab$ $initio$ density functional calculations with $DMOL^3$ package
showed that the Ni and Zn were more stable when substituting Co with formation
energy $-2.35$ eV, $2.08$ eV when Fermi level equals to the valence band
maximum. While Eu is more stable when it substitutes Na having formation energy
of $-2.64$ eV. As these results show great harmony with existing experimental
data, they provide new insights into the fundamental principle of dopant
selection for manipulating the physical properties in the development of
high-performance sodium cobaltate based multifunctional materials.
-
We develop a generalization of the Kohn-Sham density functional theory
(KS-DFT) + Hubbard $U$ (DFT+$U$) method to the excited-state regime. This has
the form of Hubbard $U$ corrected linear-response time-dependent DFT, or
`TDDFT+$U$'. Combined with calculated linear-response Hubbard $U$ parameters,
it may provide a computationally light, first-principles method for the
simulation of tightly-bound excitons on transition-metal ions. Our presented
implementation combines linear-scaling DFT+$U$ and linear-scaling TDDFT, but
the approach is broadly applicable. In detailed benchmark tests on two
Ni-centred diamagnetic coordination complexes with variable $U$ values, it is
shown that the Hubbard $U$ correction to an approximate adiabatic semi-local
exchange-correlation interaction kernel lowers the excitation energies of
transitions exclusively within the targeted localized subspace, by increasing
the exciton binding of the corresponding electron-hole pairs. This partially
counteracts the Hubbard $U$ correction to the exchange-correlation potential in
KS-DFT, which increases excitation energies into, out of, and within the
targeted localised subspace by modifying the underlying KS-DFT eigenspectrum.
This compensating effect is most pronounced for optically dark transitions
between localized orbitals of the same angular momentum, for which experimental
observation may be challenging and theoretical approaches are at their most
necessary. Overall, our results point to shortcomings in the contemporary
DFT+$U$ corrective potential, either in its functional form, or when applied to
transition-metal orbitals but not to ligand ones, or both.
-
We report studies of the correlated excited states of coronene and
substituted coronene within the Pariser-Parr-Pople (PPP) correlated
$\pi$-electron model employing symmetry adapted density matrix renormalization
group technique. These polynuclear aromatic hydrocarbons can be considered as
graphene nanoflakes. We review their electronic structures utilizing a new
symmetry adaptation scheme that exploits electron-hole symmetry, spin-inversion
symmetry and end-to-end interchange symmetry. Study of the electronic
structures sheds light on the electron correlation effects in these finite-size
graphene analogues, which diminishes on going from one-dimensional to
higher-dimensional systems, yet is significant within these finite graphene
derivatives.
-
In the present study, a new set of dimensionless coefficients is introduced
through non-dimensionalization of electrochemical equations of lead-acid
batteries. Non-dimensionalization process has been applied to the
electrochemical governing equations including conservation of charge in solid
and electrolyte, and conservation of species to derive the non-dimensional
model. Four novel dimensionless coefficients of electrode conductivity,
electrolyte conductivity, diffusional conductivity of species, and diffusion
coefficient are derived from the dimensionless model. The identified model is
validated using comparison of experimental data obtained from two lead-acid
batteries. Finally, shown results indicate that the non-dimensional model is in
fairly good accordance with data obtained from experiments, moreover,
dimensionless coefficients are useful for comparing purposes and analysis of
electrochemical processes.
-
Reversible, diffusionless, first-order solid-solid phase transitions
accompanied by caloric effects are critical for applications in the solid-state
cooling and heat-pumping devices. Accelerated discovery of caloric materials
requires reliable but faster estimators for predictions and high-throughput
screening of system-specific dominant caloric contributions. We assess
reliability of the computational methods that provide thermodynamic properties
in relevant solid phases at or near a phase transition. We test the methods
using the well-studied B2 FeRh alloy as a "fruit fly" in such a materials
genome discovery, as it exhibits a metamagnetic transition which generates
multicaloric (magneto-, elasto-, and baro-caloric) responses. For lattice
entropy contributions, we find that the commonly-used linear-response and
small-displacement phonon methods are invalid near instabilities that arise
from the anharmonicity of atomic potentials, and we offer a more reliable and
precise method for calculating lattice entropy at a fixed temperature. Then, we
apply a set of reliable methods and estimators to the metamagnetic transition
in FeRh (predicted $346 \pm 12$ K, observed $353 \pm 1$ K) and calculate the
associated caloric properties, such as isothermal entropy and isentropic
temperature changes.
-
In the present work, crystallization in melts and poor-solvent solutions of
semiflexible polymers with different concentration was studied by means of
dissipative particle dynamics simulation technique. We use a coarse-grained
polymer model trying to catch general principles of crystallization in such
systems on large time and length scales. We observe the crystallization process
starting from an initial randomly prepared system with different polymer volume
fractions in a poor solvent. Because the solvent is very poor, the macrophase
polymer-solvent separation takes place very fast and is accompanied by partial
polymer crystallization. We have found that the overall crystalline fraction at
the end of crystallization process decreases upon increasing the polymer volume
fraction in the initial randomly prepared system, while the steady-state
crystallization speed is almost the same at polymer volume fractions larger
than 50\%. At the same time, the average crystallite size differs considerably
and has a maximum value in the systems with 90\% polymer volume fraction. We
assume that this polymer concentration is an optimal value in a sense of a
balance between the amount of polymer material available for increasing
crystallite size and chain entanglements preventing crystallites growth and
merging.
-
Contrary to the conventional wisdom that deviations from standard
thermodynamics originate from the strong coupling to the bath, it is shown that
in quantum mechanics, these deviations originate from the uncertainty principle
and are supported by the non-Markovian character of the dynamics. Specifically,
it is shown that the lower bound of the dispersion of the total energy of the
system, imposed by the uncertainty principle, is dominated by the bath power
spectrum and therefore, quantum mechanics inhibits the system
thermal-equilibrium-state from being described by the canonical Boltzmann's
distribution. We show that for a wide class of systems, systems interacting via
central forces with pairwise-self-interacting environments, this general
observation is in sharp contrast to the classical case, for which the thermal
equilibrium distribution, irrespective of the interaction strength, is
\emph{exactly} characterized by the canonical Boltzmann distribution and
therefore, no dependence on the bath power spectrum is present. We define an
\emph{effective coupling} to the environment that depends on all energy scales
in the system and reservoir interaction. Sample computations in regimes
predicted by this effective coupling are demonstrated. For example, for the
case of strong effective coupling, deviations from standard thermodynamics are
present and, for the case of weak effective coupling, quantum features such as
stationary entanglement are possible at high temperatures.
-
The effect of increased electron-density (from adsorbed Li atoms) in
polyacenes and in nano-ribbons with zig-zag edge is discussed in terms of
resonance theoretical considerations and in terms edge-localized frontier
molecular orbitals. The argumentation from simple pictures is finally using the
density functional theory (DFT) for anthracene, polyacene polymer and graphene
strips. Some discussion is made for zig-zag edge graphene.
-
In this research, we employ accurate time-dependent density functional
calculations for ultrashort laser spectroscopy of nitrogen molecule. Laser
pulses with different frequencies, intensities, and durations are applied to
the molecule and the resulting photoelectron spectra are analyzed. It is argued
that relative orientation of the molecule in the laser pulse significantly
influence the orbital character of the emitted photoelectrons. Moreover, the
duration of the laser pulse is also found to be very effective in controlling
the orbital resolution and intensity of photoelectrons. Angular resolved
distribution of photoelectrons are computed at different pulse frequencies and
recording times. By exponential growth of the laser pulse intensity, the
theoretical threshold of two photons absorption in nitrogen molecule is
determined.
-
A new approach to modelling the behaviour of simple fluids is presented.
Starting from the usual expression of the partition function of N molecules, a
Fourier transformation is performed. It is argued that the N(N-1)/2 dynamical
variables kpq in the reciprocal space featuring the link between 2 molecules p
and q can reasonably be considered independent in the thermodynamical limit.
Treated as a set of effective independent particles, their statistical
behaviour is analogous to a Bose-Einstein gas. Expressions of the partition
function, of the radial pair correlation function and of the pressure are
derived, and a special attention is given to the mathematical inter-consistency
of those quantities. The results, which are independent of the exact shape of
the intermolecular potential, are applied to the simple case of hard sphere
fluids. An analytical expression of the radial pair correlation function is
derived as well as of the equation of state. The model predicts a qualitatively
satisfactory behaviour for g(R), and it provides numerically correct values for
the equation of state at low and medium densities, although at higher densities
the contact value of g(R)is underestimated. Quite interestingly it predicts for
the maximum random close packing density the correct value 0.637 .
-
Complicated mathematical equations involving products of tensors with
permutation symmetries, frequently encountered in fields such as general
relativity and quantum chemistry (e.g., equations in high-order coupled cluster
theories), require computer-based automatic derivations and manipulations. In
these processes, a key step is the collection of tensor product terms that can
be found identical by utilizing permutation symmetries of tensors or relabeling
dummy indices, which is usually achieved by defining a canonical form for
tensor product expressions. However, the problem of finding a canonical form is
nontrivial, and can be potentially of exponential cost in the number of
indices. In this work, we provided a general solution to this tensor
canonicalization problem.
-
An $\mathit{ab\ initio}$ theory is devised for the quantum dynamics of
molecules undergoing (multiple) ionization in ultrafast and intense light.
Specifically, the intertwined problem of photoionization, radiative, and
electronic transitions in the course of dissociation is addressed which arises,
e.g., when molecules are exposed to XUV light or x rays from free electron
lasers or attosecond light sources, but the approach is equally useful in
optical strong-field physics. The coherent interaction of the molecule with the
light in a specific charge state is also treated. I set out from an abstract
formulation in terms of the quantum optical notion of system-reservoir
interaction using a master equation in Lindblad form and analyze its short-time
approximation. First, I express it in a direct sum rigged Hilbert space for an
efficient solution with numerical methods for systems of differential
equations. Second, I derive a treatment via quantum Monte Carlo wave packet
(MCWP) propagation. The formalism is concretized to diatomic molecules in
Born-Oppenheimer approximation whereby molecular rotation is disregarded. The
numerical integration of the master equation is carried out with a suitably
factored density matrix that exploits the locality of the Hamiltonian and the
Lindblad superoperator with respect to the internuclear distance. The
formulation of the MCWP for molecules requires a thorough analysis of the
quantum jump process; namely, the dependence on the continuous distance renders
a straight wave packet promotion useless and, instead, a projected outer
product needs to be employed involving an integrated quantum jump operator.
-
To assist the design of novel, highly efficient molecular junctions, a deep
understanding of the precise charge transport mechanisms through these devices
is of prime importance. In the present contribution, we describe a procedure to
investigate spatially-resolved electron transport through a nanojunction from
first principles, at the example of a nitro-substituted oligo-(phenylene
ethynylene) covalently bound to graphene nanoribbon leads. Recently, we
demonstrated that the conductivity of this single-molecule-graphene-nanoribbon
junction can be switched quantitatively and reversibly upon application of a
static electric field in a top gate position, in the spirit of a traditional
field effect transistor [J. Phys. Chem. C, 2016, 120, 28808-28819]. The
propensity of the central oligomer unit to align with the external field was
found to induce a damped rotational motion and to cause an interruption of the
conjugated $\pi$-system, thereby drastically reducing the conductance through
the nanojunction. In the current work, we use the driven Liouville-von-Neumann
(DLvN) approach for time-dependent electronic transport calculations to
simulate the electronic current dynamics under time-dependent potential biases
for the two logical states of the nanojunction. Our quantum dynamical
simulations rely on a novel localization procedure using an orthonormal set of
molecular orbitals obtained from a standard density functional theory
calculation to generate a localized representation for the different parts of
the molecular junction. The transparent DLvN formalism allows us to directly
access the density matrix and to reconstruct the time-dependent electronic
current density, unraveling unique mechanistic details of the electron
transport.
-
We report extensive theoretical calculations on the rotation-inversion
excitation of interstellar ammonia (NH3) due to collisions with atomic and
molecular hydrogen (both para- and ortho-H2). Close-coupling calculations are
performed for total energies in the range 1-2000 cm-1 and rotational cross
sections are obtained for all transitions among the lowest 17 and 34
rotation-inversion levels of ortho- and para-NH3, respectively. Rate
coefficients are deduced for kinetic temperatures up to 200 K. Propensity rules
for the three colliding partners are discussed and we also compare the new
results to previous calculations for the spherically symmetrical He and para-H2
projectiles. Significant differences are found between the different sets of
calculations. Finally, we test the impact of the new rate coefficients on the
calibration of the ammonia thermometer. We find that the calibration curve is
only weakly sensitive to the colliding partner and we confirm that the ammonia
thermometer is robust.
-
Upon hydrogen bond formation, electronic charge density is transferred
between the donor and acceptor, impacting processes ranging from hydration to
spectroscopy. Here we use ab initio path integral simulations to elucidate the
role of nuclear quantum effects in determining the charge transfer in a range
of hydrogen bonded species in the gas and liquid phase. We show that the
quantization of the nuclei gives rise to large changes in the magnitude of the
charge transfer as well as its temperature dependence. We then explain how a
single geometric parameter determines the charge transfer through the hydrogen
bond. These results thus demonstrate that nuclear quantum effects are vital for
the accurate description of charge transfer and offer a physically transparent
way to understand how hydrogen bonding gives rise to it.





 Photoisomerization in a system with multiple electronic states and anharmonic potential surfaces in a dissipative environment is investigated using a rigorous numerical method employing quantum hierarchical Fokker-Planck equations (QHFPE) for multi-state systems. We have developed a computer code incorporating QHFPE for general-purpose computing on graphics processing units (GPGPU) that can treat multi-state systems in phase space with any strength of diabatic coupling of electronic states under non-perturbative and non-Markovian system-bath interactions. This approach facilitates the calculation of both linear and nonlinear spectra. We computed Wigner distributions for excited, ground, and coherent states. We then investigated excited state dynamics involving transitions among these states by analyzing linear absorption and transient absorption processes and multi-dimensional electronic spectra with various values of the heat bath parameters. Our results provide predictions for spectroscopic measurements of photoisomerization dynamics. The motion of excitation and ground state wavepackets and their coherence involved in the photoisomerization were observed as the profiles of positive and negative peaks of two-dimensional spectra.
Photoisomerization in a system with multiple electronic states and anharmonic potential surfaces in a dissipative environment is investigated using a rigorous numerical method employing quantum hierarchical Fokker-Planck equations (QHFPE) for multi-state systems. We have developed a computer code incorporating QHFPE for general-purpose computing on graphics processing units (GPGPU) that can treat multi-state systems in phase space with any strength of diabatic coupling of electronic states under non-perturbative and non-Markovian system-bath interactions. This approach facilitates the calculation of both linear and nonlinear spectra. We computed Wigner distributions for excited, ground, and coherent states. We then investigated excited state dynamics involving transitions among these states by analyzing linear absorption and transient absorption processes and multi-dimensional electronic spectra with various values of the heat bath parameters. Our results provide predictions for spectroscopic measurements of photoisomerization dynamics. The motion of excitation and ground state wavepackets and their coherence involved in the photoisomerization were observed as the profiles of positive and negative peaks of two-dimensional spectra.
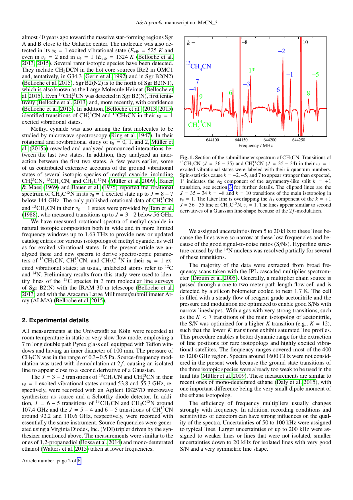



 Methyl cyanide is an important trace molecule in space, especially in star-forming regions where it is one of the more common molecules used to derive kinetic temperatures. We want to obtain accurate spectroscopic parameters of minor isotopologs of methyl cyanide in their lowest excited $v_8 = 1$ vibrational states to support astronomical observations, in particular, with interferometers such as ALMA. The laboratory rotational spectrum of methyl cyanide in natural isotopic composition has been recorded from the millimeter to the terahertz regions. Transitions with good signal-to-noise ratios could be identified for the three isotopic species CH$_3^{13}$CN, $^{13}$CH$_3$CN, and CH$_3$C(15)N up to about 1.2 THz ($J'' \le 66$). Accurate spectroscopic parameters were obtained for all three species. The present data were already instrumental in identifying $v_8 = 1$ lines of methyl cyanide with one $^{13}$C in IRAM 30 m and ALMA data toward Sagittarius B2(N).
Methyl cyanide is an important trace molecule in space, especially in star-forming regions where it is one of the more common molecules used to derive kinetic temperatures. We want to obtain accurate spectroscopic parameters of minor isotopologs of methyl cyanide in their lowest excited $v_8 = 1$ vibrational states to support astronomical observations, in particular, with interferometers such as ALMA. The laboratory rotational spectrum of methyl cyanide in natural isotopic composition has been recorded from the millimeter to the terahertz regions. Transitions with good signal-to-noise ratios could be identified for the three isotopic species CH$_3^{13}$CN, $^{13}$CH$_3$CN, and CH$_3$C(15)N up to about 1.2 THz ($J'' \le 66$). Accurate spectroscopic parameters were obtained for all three species. The present data were already instrumental in identifying $v_8 = 1$ lines of methyl cyanide with one $^{13}$C in IRAM 30 m and ALMA data toward Sagittarius B2(N).




 Ligand diffusion through proteins is a fundamental process governing biological signaling and enzymatic catalysis. The complex topology of protein tunnels results in difficulties with computing ligand escape pathways by standard molecular dynamics (MD) simulations. Here, two novel methods for searching of ligand exit pathways and cavity exploration are proposed: memory random acceleration MD (mRAMD), and memetic algorithms (MA). In mRAMD, finding exit pathways is based on a non-Markovian biasing that is introduced to optimize the unbinding force. In MA, hybrid learning protocols are exploited to predict optimal ligand exit paths. The methods are tested on three proteins with increasing complexity of tunnels: M2 muscarinic receptor, nitrile hydratase, and cytochrome P450cam. In these cases, the proposed methods outperform standard techniques that are used currently to find ligand egress pathways. The proposed approach is general and appropriate for accelerated transport of an object through a network of protein tunnels.
Ligand diffusion through proteins is a fundamental process governing biological signaling and enzymatic catalysis. The complex topology of protein tunnels results in difficulties with computing ligand escape pathways by standard molecular dynamics (MD) simulations. Here, two novel methods for searching of ligand exit pathways and cavity exploration are proposed: memory random acceleration MD (mRAMD), and memetic algorithms (MA). In mRAMD, finding exit pathways is based on a non-Markovian biasing that is introduced to optimize the unbinding force. In MA, hybrid learning protocols are exploited to predict optimal ligand exit paths. The methods are tested on three proteins with increasing complexity of tunnels: M2 muscarinic receptor, nitrile hydratase, and cytochrome P450cam. In these cases, the proposed methods outperform standard techniques that are used currently to find ligand egress pathways. The proposed approach is general and appropriate for accelerated transport of an object through a network of protein tunnels.

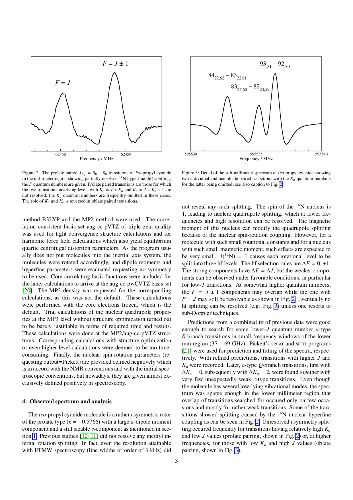
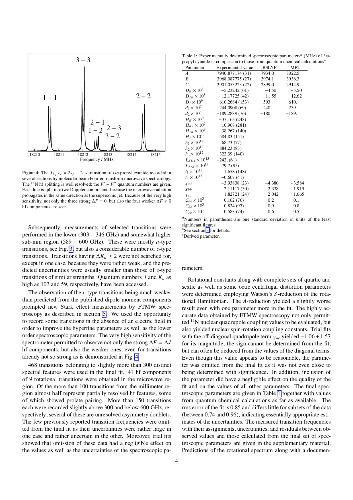
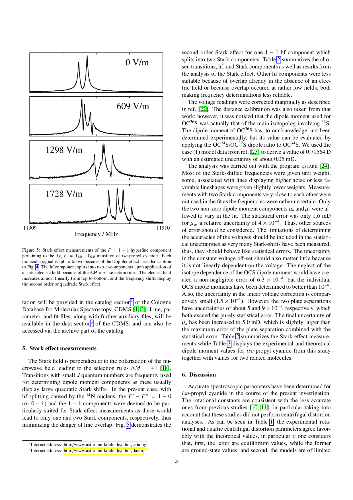
 Rotational transitions of $iso$-propyl cyanide, (CH$_3$)$_2$CHCN, also known as $iso$-butyronitrile, were recorded using long-path absorption spectroscopy in selected regions between 37 and 600 GHz. Further measurements were carried out between 6 and 20 GHz employing Fourier transform microwave (FTMW) spectroscopy on a pulsed molecular supersonic jet. The observed transitions reach $J$ and $K_a$ quantum numbers of 103 and 59, respectively, and yield accurate rotational constants as well as distortion parameters up to eighth order. The $^{14}$N nuclear hyperfine splitting was resolved in particular by FTMW spectroscopy yielding spin-rotation parameters as well as very accurate quadrupole coupling terms. In addition, Stark effect measurements were carried out in the microwave region to obtain a largely revised $c$-dipole moment component and to improve the $a$-component. The hyperfine coupling and dipole moment values are compared with values for related molecules both from experiment and from quantum chemical calculations.
Rotational transitions of $iso$-propyl cyanide, (CH$_3$)$_2$CHCN, also known as $iso$-butyronitrile, were recorded using long-path absorption spectroscopy in selected regions between 37 and 600 GHz. Further measurements were carried out between 6 and 20 GHz employing Fourier transform microwave (FTMW) spectroscopy on a pulsed molecular supersonic jet. The observed transitions reach $J$ and $K_a$ quantum numbers of 103 and 59, respectively, and yield accurate rotational constants as well as distortion parameters up to eighth order. The $^{14}$N nuclear hyperfine splitting was resolved in particular by FTMW spectroscopy yielding spin-rotation parameters as well as very accurate quadrupole coupling terms. In addition, Stark effect measurements were carried out in the microwave region to obtain a largely revised $c$-dipole moment component and to improve the $a$-component. The hyperfine coupling and dipole moment values are compared with values for related molecules both from experiment and from quantum chemical calculations.


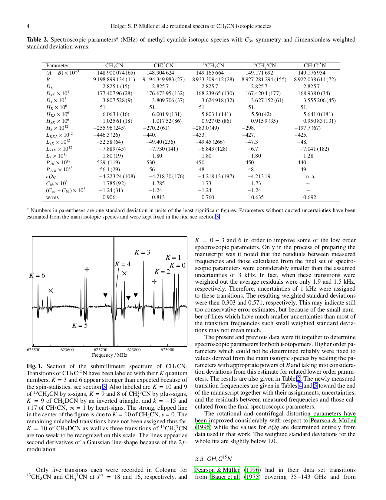

 Methyl cyanide is an important trace molecule in star-forming regions. It is one of the more common molecules used to derive kinetic temperatures in such sources. As preparatory work for Herschel, SOFIA, and in particular ALMA we want to improve the rest frequencies of the main as well as minor isotopologs of methyl cyanide. The laboratory rotational spectrum of methyl cyanide in natural isotopic composition has been recorded up to 1.63 THz. Transitions with good signal-to-noise ratio could be identified for CH$_3$CN, $^{13}$CH$_3$CN, CH$_3^{13}$CN, CH$_3$C$^{15}$N, CH$_2$DCN, and $^{13}$CH$_3^{13}$CN in their ground vibrational states up to about 1.2 THz. The main isotopic species could be identified even in the highest frequency spectral recordings around 1.6 THz. The highest $J'$ quantum numbers included in the fit are 64 for $^{13}$CH$_3^{13}$CN and 89 for the main isotopic species. Greatly improved spectroscopic parameters have been obtained by fitting the present data together with previously reported transition frequencies. The present data will be helpful to identify isotopologs of methyl cyanide in the higher frequency bands of instruments such as the recently launched Herschel satellite, the upcoming airplane mission SOFIA or the radio telescope array ALMA.
Methyl cyanide is an important trace molecule in star-forming regions. It is one of the more common molecules used to derive kinetic temperatures in such sources. As preparatory work for Herschel, SOFIA, and in particular ALMA we want to improve the rest frequencies of the main as well as minor isotopologs of methyl cyanide. The laboratory rotational spectrum of methyl cyanide in natural isotopic composition has been recorded up to 1.63 THz. Transitions with good signal-to-noise ratio could be identified for CH$_3$CN, $^{13}$CH$_3$CN, CH$_3^{13}$CN, CH$_3$C$^{15}$N, CH$_2$DCN, and $^{13}$CH$_3^{13}$CN in their ground vibrational states up to about 1.2 THz. The main isotopic species could be identified even in the highest frequency spectral recordings around 1.6 THz. The highest $J'$ quantum numbers included in the fit are 64 for $^{13}$CH$_3^{13}$CN and 89 for the main isotopic species. Greatly improved spectroscopic parameters have been obtained by fitting the present data together with previously reported transition frequencies. The present data will be helpful to identify isotopologs of methyl cyanide in the higher frequency bands of instruments such as the recently launched Herschel satellite, the upcoming airplane mission SOFIA or the radio telescope array ALMA.


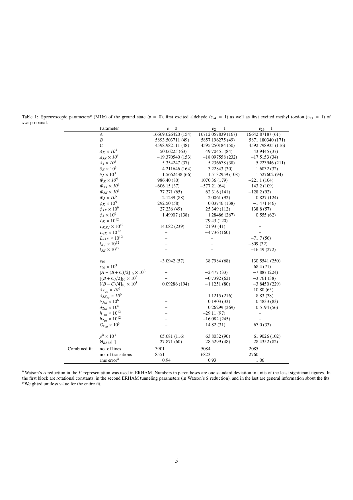
 The rotational spectra of the two stable conformers syn- and gauche-propanal (CH$_3$CH$_2$CHO) were studied in the millimeter and submillimeter wave regions from 75 to 500 GHz with the Cologne (Sub-)Millimeter wave Spectrometer. Furthermore, the first excited states associated with the aldehyde torsion and with the methyl torsion, respectively, of the $syn$-conformer were analyzed. The newly obtained spectroscopic parameters yield better predictions, thus fulfill sensitivity and resolution requirements in new astronomical observations in order to unambiguously assign pure rotational transitions of propanal. This is demonstrated on a radio astronomical spectrum from the Atacama Large Millimeter/submillimeter Array Protostellar Interferometric Line Survey (ALMA-PILS). In particular, an accurate description of observed splittings, caused by internal rotation of the methyl group in the $syn$-conformer and by tunneling rotation interaction from two stable degenerate $gauche$-conformers, is reported. The rotational spectrum of propanal is of additional interest because of its two large amplitude motions pertaining to the methyl and the aldehyde group, respectively.
The rotational spectra of the two stable conformers syn- and gauche-propanal (CH$_3$CH$_2$CHO) were studied in the millimeter and submillimeter wave regions from 75 to 500 GHz with the Cologne (Sub-)Millimeter wave Spectrometer. Furthermore, the first excited states associated with the aldehyde torsion and with the methyl torsion, respectively, of the $syn$-conformer were analyzed. The newly obtained spectroscopic parameters yield better predictions, thus fulfill sensitivity and resolution requirements in new astronomical observations in order to unambiguously assign pure rotational transitions of propanal. This is demonstrated on a radio astronomical spectrum from the Atacama Large Millimeter/submillimeter Array Protostellar Interferometric Line Survey (ALMA-PILS). In particular, an accurate description of observed splittings, caused by internal rotation of the methyl group in the $syn$-conformer and by tunneling rotation interaction from two stable degenerate $gauche$-conformers, is reported. The rotational spectrum of propanal is of additional interest because of its two large amplitude motions pertaining to the methyl and the aldehyde group, respectively.


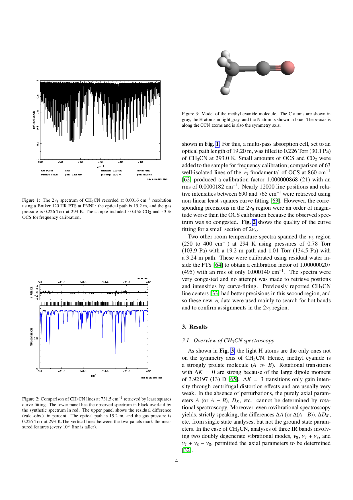

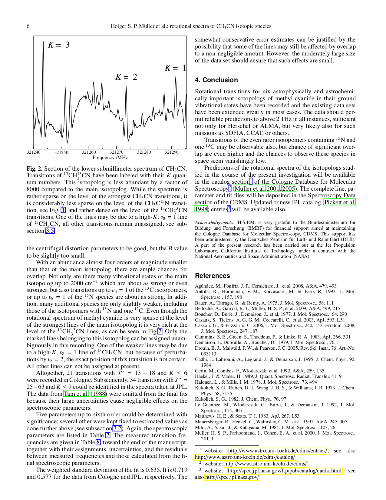
 Spectra of methyl cyanide were recorded to analyze interactions in low-lying vibrational states and to construct line lists for radio astronomical observations as well as for infrared spectroscopic investigations of planetary atmospheres. The rotational spectra cover large portions of the 36$-$1627 GHz region. In the infrared (IR), a spectrum was recorded for this study in the region of 2$\nu _8$ around 717 cm$^{-1}$ with assignments covering 684$-$765 cm$^{-1}$. Additional spectra in the $\nu _8$ region were used to validate the analysis. The large amount and the high accuracy of the rotational data extend to much higher $J$ and $K$ quantum numbers and allowed us to investigate for the first time in depth local interactions between these states which occur at high $K$ values. In particular, we have detected several interactions between $v_8 = 1$ and 2. Notably, there is a strong $\Delta v_8 = \pm1$, $\Delta K = 0$, $\Delta l = \pm3$ Fermi resonance between $v_8 = 1^{-1}$ and $v_8 = 2^{+2}$ at $K$ = 14. Pronounced effects in the spectrum are also caused by resonant $\Delta v_8 = \pm1$, $\Delta K = \mp2$, $\Delta l = \pm1$ interactions between $v_8 = 1$ and 2. An equivalent resonant interaction occurs between $K$ = 14 of the ground vibrational state and $K$ = 12, $l = +1$ of $v_8 = 1$ for which we present the first detailed account. A preliminary account was given in an earlier study on the ground vibrational state. From data pertaining to $v_8 = 2$, we also investigated rotational interactions with $v_4 = 1$ as well as $\Delta v_8 = \pm1$, $\Delta K = 0$, $\Delta l = \pm3$ Fermi interactions between $v_8 = 2$ and 3. We have derived N$_2$- and self-broadening coefficients for the $\nu _8$, 2$\nu _8 - \nu _8$, and 2$\nu _8$ bands from previously determined nu4 values. Subsequently, we determined transition moments and intensities for the three IR bands.
Spectra of methyl cyanide were recorded to analyze interactions in low-lying vibrational states and to construct line lists for radio astronomical observations as well as for infrared spectroscopic investigations of planetary atmospheres. The rotational spectra cover large portions of the 36$-$1627 GHz region. In the infrared (IR), a spectrum was recorded for this study in the region of 2$\nu _8$ around 717 cm$^{-1}$ with assignments covering 684$-$765 cm$^{-1}$. Additional spectra in the $\nu _8$ region were used to validate the analysis. The large amount and the high accuracy of the rotational data extend to much higher $J$ and $K$ quantum numbers and allowed us to investigate for the first time in depth local interactions between these states which occur at high $K$ values. In particular, we have detected several interactions between $v_8 = 1$ and 2. Notably, there is a strong $\Delta v_8 = \pm1$, $\Delta K = 0$, $\Delta l = \pm3$ Fermi resonance between $v_8 = 1^{-1}$ and $v_8 = 2^{+2}$ at $K$ = 14. Pronounced effects in the spectrum are also caused by resonant $\Delta v_8 = \pm1$, $\Delta K = \mp2$, $\Delta l = \pm1$ interactions between $v_8 = 1$ and 2. An equivalent resonant interaction occurs between $K$ = 14 of the ground vibrational state and $K$ = 12, $l = +1$ of $v_8 = 1$ for which we present the first detailed account. A preliminary account was given in an earlier study on the ground vibrational state. From data pertaining to $v_8 = 2$, we also investigated rotational interactions with $v_4 = 1$ as well as $\Delta v_8 = \pm1$, $\Delta K = 0$, $\Delta l = \pm3$ Fermi interactions between $v_8 = 2$ and 3. We have derived N$_2$- and self-broadening coefficients for the $\nu _8$, 2$\nu _8 - \nu _8$, and 2$\nu _8$ bands from previously determined nu4 values. Subsequently, we determined transition moments and intensities for the three IR bands.

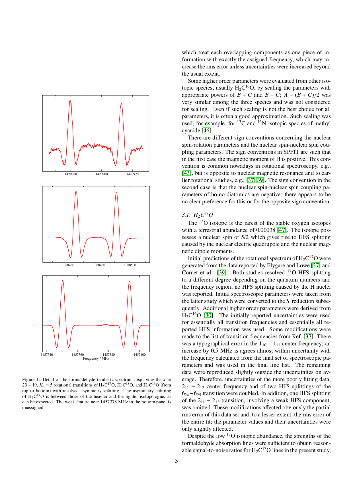


 The rotational spectrum of the formaldehyde isotopologue H$_2$C$^{17}$O was investigated between 0.56 and 1.50 THz using a sample of natural isotopic composition. In addition, transition frequencies were determined for H$_2$C$^{18}$O and H$_2$C$^{16}$O between 1.37 and 1.50 THz. The data were combined with critically evaluated literature data to derive improved sets of spectroscopic parameters which include $^{17}$O or H nuclear hyperfine structure parameters.
The rotational spectrum of the formaldehyde isotopologue H$_2$C$^{17}$O was investigated between 0.56 and 1.50 THz using a sample of natural isotopic composition. In addition, transition frequencies were determined for H$_2$C$^{18}$O and H$_2$C$^{16}$O between 1.37 and 1.50 THz. The data were combined with critically evaluated literature data to derive improved sets of spectroscopic parameters which include $^{17}$O or H nuclear hyperfine structure parameters.




 We present a new state function, denotes the energy of thermal motion, it shows that the energy classification for the internal energy has been completed. In a new theoretical structure, we present the concept of the heat partial pressure, rewrite the equations of the first law, by which, the energy conversion and energy transport can be distinguished by an explicit way, we redefine the concept of the entropy, and confirm a conversion potential related to the dissipative compensation, we discuss in detail on the three sources of irreversibility, and the conversions between the calorimetric entropy and the configurational entropy, an interesting conclusion shows that the second law itself has contained the mechanism of evolution.
We present a new state function, denotes the energy of thermal motion, it shows that the energy classification for the internal energy has been completed. In a new theoretical structure, we present the concept of the heat partial pressure, rewrite the equations of the first law, by which, the energy conversion and energy transport can be distinguished by an explicit way, we redefine the concept of the entropy, and confirm a conversion potential related to the dissipative compensation, we discuss in detail on the three sources of irreversibility, and the conversions between the calorimetric entropy and the configurational entropy, an interesting conclusion shows that the second law itself has contained the mechanism of evolution.

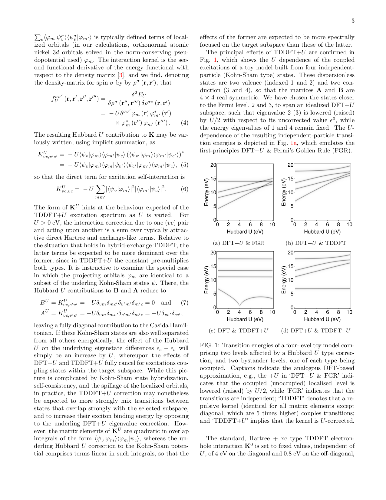
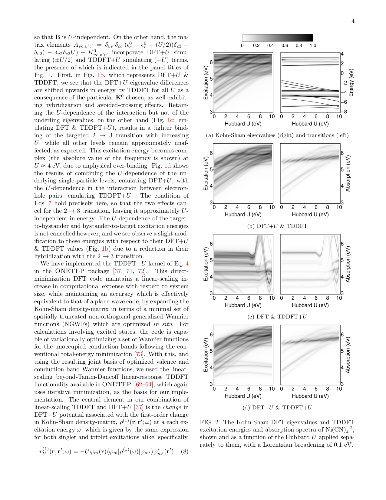
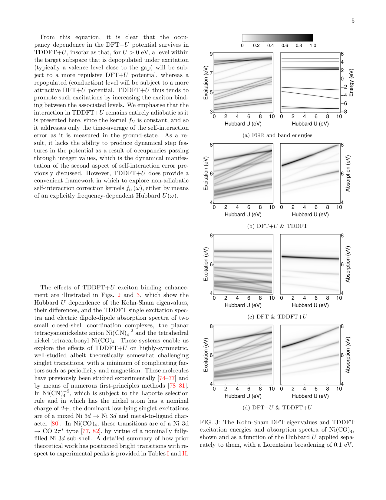
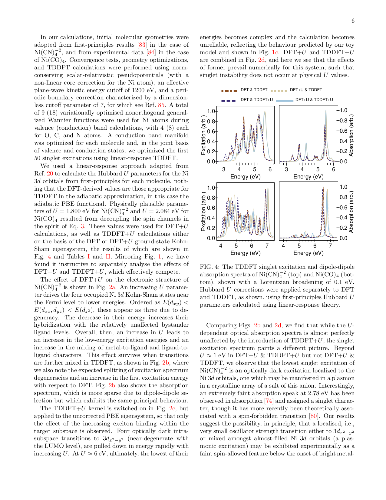 We develop a generalization of the Kohn-Sham density functional theory (KS-DFT) + Hubbard $U$ (DFT+$U$) method to the excited-state regime. This has the form of Hubbard $U$ corrected linear-response time-dependent DFT, or `TDDFT+$U$'. Combined with calculated linear-response Hubbard $U$ parameters, it may provide a computationally light, first-principles method for the simulation of tightly-bound excitons on transition-metal ions. Our presented implementation combines linear-scaling DFT+$U$ and linear-scaling TDDFT, but the approach is broadly applicable. In detailed benchmark tests on two Ni-centred diamagnetic coordination complexes with variable $U$ values, it is shown that the Hubbard $U$ correction to an approximate adiabatic semi-local exchange-correlation interaction kernel lowers the excitation energies of transitions exclusively within the targeted localized subspace, by increasing the exciton binding of the corresponding electron-hole pairs. This partially counteracts the Hubbard $U$ correction to the exchange-correlation potential in KS-DFT, which increases excitation energies into, out of, and within the targeted localised subspace by modifying the underlying KS-DFT eigenspectrum. This compensating effect is most pronounced for optically dark transitions between localized orbitals of the same angular momentum, for which experimental observation may be challenging and theoretical approaches are at their most necessary. Overall, our results point to shortcomings in the contemporary DFT+$U$ corrective potential, either in its functional form, or when applied to transition-metal orbitals but not to ligand ones, or both.
We develop a generalization of the Kohn-Sham density functional theory (KS-DFT) + Hubbard $U$ (DFT+$U$) method to the excited-state regime. This has the form of Hubbard $U$ corrected linear-response time-dependent DFT, or `TDDFT+$U$'. Combined with calculated linear-response Hubbard $U$ parameters, it may provide a computationally light, first-principles method for the simulation of tightly-bound excitons on transition-metal ions. Our presented implementation combines linear-scaling DFT+$U$ and linear-scaling TDDFT, but the approach is broadly applicable. In detailed benchmark tests on two Ni-centred diamagnetic coordination complexes with variable $U$ values, it is shown that the Hubbard $U$ correction to an approximate adiabatic semi-local exchange-correlation interaction kernel lowers the excitation energies of transitions exclusively within the targeted localized subspace, by increasing the exciton binding of the corresponding electron-hole pairs. This partially counteracts the Hubbard $U$ correction to the exchange-correlation potential in KS-DFT, which increases excitation energies into, out of, and within the targeted localised subspace by modifying the underlying KS-DFT eigenspectrum. This compensating effect is most pronounced for optically dark transitions between localized orbitals of the same angular momentum, for which experimental observation may be challenging and theoretical approaches are at their most necessary. Overall, our results point to shortcomings in the contemporary DFT+$U$ corrective potential, either in its functional form, or when applied to transition-metal orbitals but not to ligand ones, or both.




 We report studies of the correlated excited states of coronene and substituted coronene within the Pariser-Parr-Pople (PPP) correlated $\pi$-electron model employing symmetry adapted density matrix renormalization group technique. These polynuclear aromatic hydrocarbons can be considered as graphene nanoflakes. We review their electronic structures utilizing a new symmetry adaptation scheme that exploits electron-hole symmetry, spin-inversion symmetry and end-to-end interchange symmetry. Study of the electronic structures sheds light on the electron correlation effects in these finite-size graphene analogues, which diminishes on going from one-dimensional to higher-dimensional systems, yet is significant within these finite graphene derivatives.
We report studies of the correlated excited states of coronene and substituted coronene within the Pariser-Parr-Pople (PPP) correlated $\pi$-electron model employing symmetry adapted density matrix renormalization group technique. These polynuclear aromatic hydrocarbons can be considered as graphene nanoflakes. We review their electronic structures utilizing a new symmetry adaptation scheme that exploits electron-hole symmetry, spin-inversion symmetry and end-to-end interchange symmetry. Study of the electronic structures sheds light on the electron correlation effects in these finite-size graphene analogues, which diminishes on going from one-dimensional to higher-dimensional systems, yet is significant within these finite graphene derivatives.




 In the present study, a new set of dimensionless coefficients is introduced through non-dimensionalization of electrochemical equations of lead-acid batteries. Non-dimensionalization process has been applied to the electrochemical governing equations including conservation of charge in solid and electrolyte, and conservation of species to derive the non-dimensional model. Four novel dimensionless coefficients of electrode conductivity, electrolyte conductivity, diffusional conductivity of species, and diffusion coefficient are derived from the dimensionless model. The identified model is validated using comparison of experimental data obtained from two lead-acid batteries. Finally, shown results indicate that the non-dimensional model is in fairly good accordance with data obtained from experiments, moreover, dimensionless coefficients are useful for comparing purposes and analysis of electrochemical processes.
In the present study, a new set of dimensionless coefficients is introduced through non-dimensionalization of electrochemical equations of lead-acid batteries. Non-dimensionalization process has been applied to the electrochemical governing equations including conservation of charge in solid and electrolyte, and conservation of species to derive the non-dimensional model. Four novel dimensionless coefficients of electrode conductivity, electrolyte conductivity, diffusional conductivity of species, and diffusion coefficient are derived from the dimensionless model. The identified model is validated using comparison of experimental data obtained from two lead-acid batteries. Finally, shown results indicate that the non-dimensional model is in fairly good accordance with data obtained from experiments, moreover, dimensionless coefficients are useful for comparing purposes and analysis of electrochemical processes.
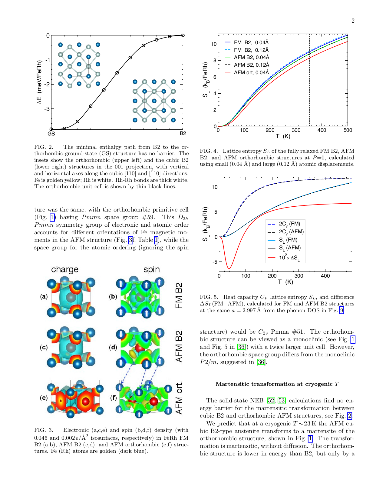
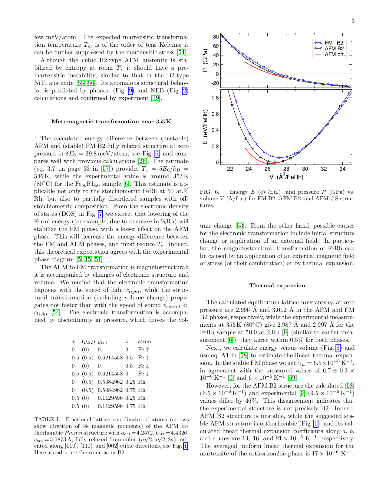
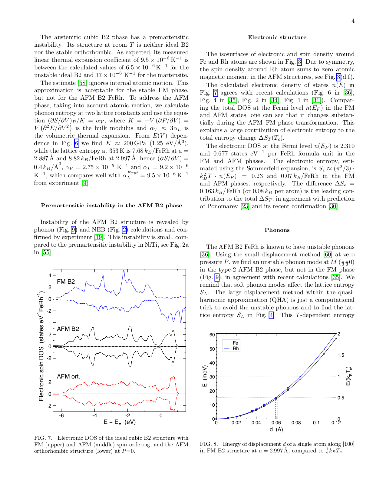
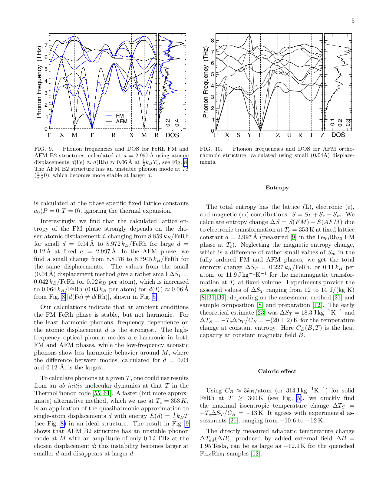
 Reversible, diffusionless, first-order solid-solid phase transitions accompanied by caloric effects are critical for applications in the solid-state cooling and heat-pumping devices. Accelerated discovery of caloric materials requires reliable but faster estimators for predictions and high-throughput screening of system-specific dominant caloric contributions. We assess reliability of the computational methods that provide thermodynamic properties in relevant solid phases at or near a phase transition. We test the methods using the well-studied B2 FeRh alloy as a "fruit fly" in such a materials genome discovery, as it exhibits a metamagnetic transition which generates multicaloric (magneto-, elasto-, and baro-caloric) responses. For lattice entropy contributions, we find that the commonly-used linear-response and small-displacement phonon methods are invalid near instabilities that arise from the anharmonicity of atomic potentials, and we offer a more reliable and precise method for calculating lattice entropy at a fixed temperature. Then, we apply a set of reliable methods and estimators to the metamagnetic transition in FeRh (predicted $346 \pm 12$ K, observed $353 \pm 1$ K) and calculate the associated caloric properties, such as isothermal entropy and isentropic temperature changes.
Reversible, diffusionless, first-order solid-solid phase transitions accompanied by caloric effects are critical for applications in the solid-state cooling and heat-pumping devices. Accelerated discovery of caloric materials requires reliable but faster estimators for predictions and high-throughput screening of system-specific dominant caloric contributions. We assess reliability of the computational methods that provide thermodynamic properties in relevant solid phases at or near a phase transition. We test the methods using the well-studied B2 FeRh alloy as a "fruit fly" in such a materials genome discovery, as it exhibits a metamagnetic transition which generates multicaloric (magneto-, elasto-, and baro-caloric) responses. For lattice entropy contributions, we find that the commonly-used linear-response and small-displacement phonon methods are invalid near instabilities that arise from the anharmonicity of atomic potentials, and we offer a more reliable and precise method for calculating lattice entropy at a fixed temperature. Then, we apply a set of reliable methods and estimators to the metamagnetic transition in FeRh (predicted $346 \pm 12$ K, observed $353 \pm 1$ K) and calculate the associated caloric properties, such as isothermal entropy and isentropic temperature changes.




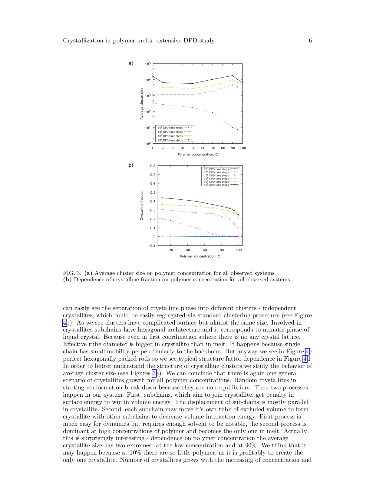 In the present work, crystallization in melts and poor-solvent solutions of semiflexible polymers with different concentration was studied by means of dissipative particle dynamics simulation technique. We use a coarse-grained polymer model trying to catch general principles of crystallization in such systems on large time and length scales. We observe the crystallization process starting from an initial randomly prepared system with different polymer volume fractions in a poor solvent. Because the solvent is very poor, the macrophase polymer-solvent separation takes place very fast and is accompanied by partial polymer crystallization. We have found that the overall crystalline fraction at the end of crystallization process decreases upon increasing the polymer volume fraction in the initial randomly prepared system, while the steady-state crystallization speed is almost the same at polymer volume fractions larger than 50\%. At the same time, the average crystallite size differs considerably and has a maximum value in the systems with 90\% polymer volume fraction. We assume that this polymer concentration is an optimal value in a sense of a balance between the amount of polymer material available for increasing crystallite size and chain entanglements preventing crystallites growth and merging.
In the present work, crystallization in melts and poor-solvent solutions of semiflexible polymers with different concentration was studied by means of dissipative particle dynamics simulation technique. We use a coarse-grained polymer model trying to catch general principles of crystallization in such systems on large time and length scales. We observe the crystallization process starting from an initial randomly prepared system with different polymer volume fractions in a poor solvent. Because the solvent is very poor, the macrophase polymer-solvent separation takes place very fast and is accompanied by partial polymer crystallization. We have found that the overall crystalline fraction at the end of crystallization process decreases upon increasing the polymer volume fraction in the initial randomly prepared system, while the steady-state crystallization speed is almost the same at polymer volume fractions larger than 50\%. At the same time, the average crystallite size differs considerably and has a maximum value in the systems with 90\% polymer volume fraction. We assume that this polymer concentration is an optimal value in a sense of a balance between the amount of polymer material available for increasing crystallite size and chain entanglements preventing crystallites growth and merging.



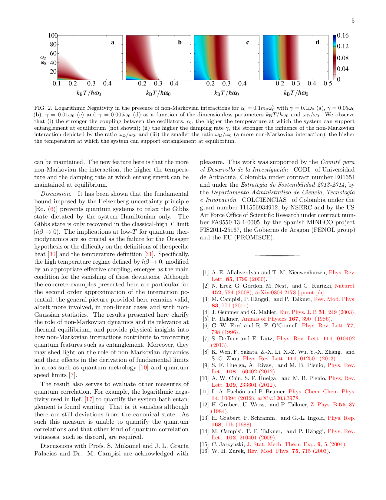
 Contrary to the conventional wisdom that deviations from standard thermodynamics originate from the strong coupling to the bath, it is shown that in quantum mechanics, these deviations originate from the uncertainty principle and are supported by the non-Markovian character of the dynamics. Specifically, it is shown that the lower bound of the dispersion of the total energy of the system, imposed by the uncertainty principle, is dominated by the bath power spectrum and therefore, quantum mechanics inhibits the system thermal-equilibrium-state from being described by the canonical Boltzmann's distribution. We show that for a wide class of systems, systems interacting via central forces with pairwise-self-interacting environments, this general observation is in sharp contrast to the classical case, for which the thermal equilibrium distribution, irrespective of the interaction strength, is \emph{exactly} characterized by the canonical Boltzmann distribution and therefore, no dependence on the bath power spectrum is present. We define an \emph{effective coupling} to the environment that depends on all energy scales in the system and reservoir interaction. Sample computations in regimes predicted by this effective coupling are demonstrated. For example, for the case of strong effective coupling, deviations from standard thermodynamics are present and, for the case of weak effective coupling, quantum features such as stationary entanglement are possible at high temperatures.
Contrary to the conventional wisdom that deviations from standard thermodynamics originate from the strong coupling to the bath, it is shown that in quantum mechanics, these deviations originate from the uncertainty principle and are supported by the non-Markovian character of the dynamics. Specifically, it is shown that the lower bound of the dispersion of the total energy of the system, imposed by the uncertainty principle, is dominated by the bath power spectrum and therefore, quantum mechanics inhibits the system thermal-equilibrium-state from being described by the canonical Boltzmann's distribution. We show that for a wide class of systems, systems interacting via central forces with pairwise-self-interacting environments, this general observation is in sharp contrast to the classical case, for which the thermal equilibrium distribution, irrespective of the interaction strength, is \emph{exactly} characterized by the canonical Boltzmann distribution and therefore, no dependence on the bath power spectrum is present. We define an \emph{effective coupling} to the environment that depends on all energy scales in the system and reservoir interaction. Sample computations in regimes predicted by this effective coupling are demonstrated. For example, for the case of strong effective coupling, deviations from standard thermodynamics are present and, for the case of weak effective coupling, quantum features such as stationary entanglement are possible at high temperatures.


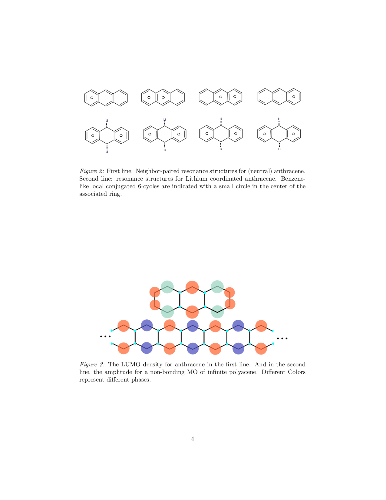

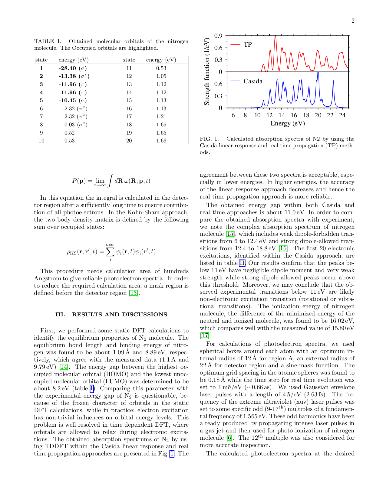
 The effect of increased electron-density (from adsorbed Li atoms) in polyacenes and in nano-ribbons with zig-zag edge is discussed in terms of resonance theoretical considerations and in terms edge-localized frontier molecular orbitals. The argumentation from simple pictures is finally using the density functional theory (DFT) for anthracene, polyacene polymer and graphene strips. Some discussion is made for zig-zag edge graphene.
The effect of increased electron-density (from adsorbed Li atoms) in polyacenes and in nano-ribbons with zig-zag edge is discussed in terms of resonance theoretical considerations and in terms edge-localized frontier molecular orbitals. The argumentation from simple pictures is finally using the density functional theory (DFT) for anthracene, polyacene polymer and graphene strips. Some discussion is made for zig-zag edge graphene.
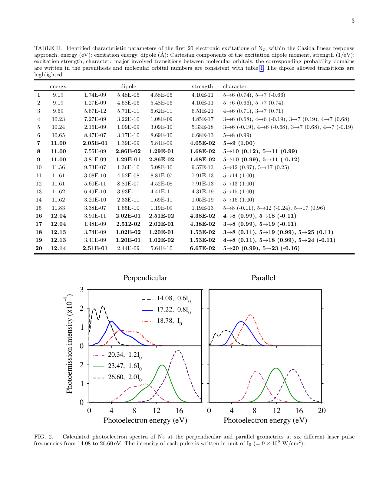
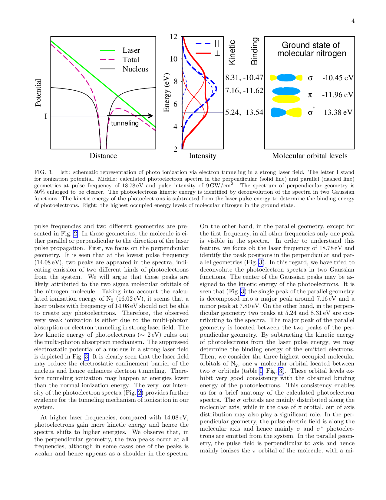
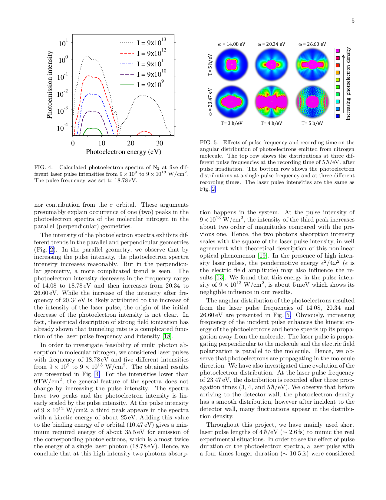
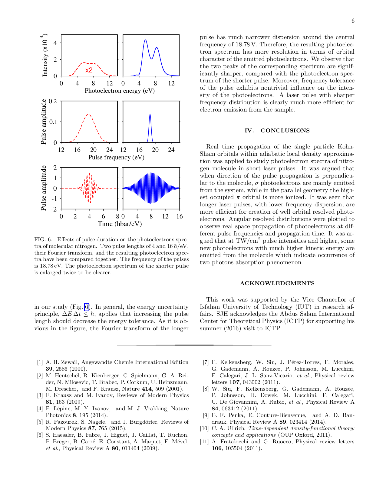 In this research, we employ accurate time-dependent density functional calculations for ultrashort laser spectroscopy of nitrogen molecule. Laser pulses with different frequencies, intensities, and durations are applied to the molecule and the resulting photoelectron spectra are analyzed. It is argued that relative orientation of the molecule in the laser pulse significantly influence the orbital character of the emitted photoelectrons. Moreover, the duration of the laser pulse is also found to be very effective in controlling the orbital resolution and intensity of photoelectrons. Angular resolved distribution of photoelectrons are computed at different pulse frequencies and recording times. By exponential growth of the laser pulse intensity, the theoretical threshold of two photons absorption in nitrogen molecule is determined.
In this research, we employ accurate time-dependent density functional calculations for ultrashort laser spectroscopy of nitrogen molecule. Laser pulses with different frequencies, intensities, and durations are applied to the molecule and the resulting photoelectron spectra are analyzed. It is argued that relative orientation of the molecule in the laser pulse significantly influence the orbital character of the emitted photoelectrons. Moreover, the duration of the laser pulse is also found to be very effective in controlling the orbital resolution and intensity of photoelectrons. Angular resolved distribution of photoelectrons are computed at different pulse frequencies and recording times. By exponential growth of the laser pulse intensity, the theoretical threshold of two photons absorption in nitrogen molecule is determined.




 Complicated mathematical equations involving products of tensors with permutation symmetries, frequently encountered in fields such as general relativity and quantum chemistry (e.g., equations in high-order coupled cluster theories), require computer-based automatic derivations and manipulations. In these processes, a key step is the collection of tensor product terms that can be found identical by utilizing permutation symmetries of tensors or relabeling dummy indices, which is usually achieved by defining a canonical form for tensor product expressions. However, the problem of finding a canonical form is nontrivial, and can be potentially of exponential cost in the number of indices. In this work, we provided a general solution to this tensor canonicalization problem.
Complicated mathematical equations involving products of tensors with permutation symmetries, frequently encountered in fields such as general relativity and quantum chemistry (e.g., equations in high-order coupled cluster theories), require computer-based automatic derivations and manipulations. In these processes, a key step is the collection of tensor product terms that can be found identical by utilizing permutation symmetries of tensors or relabeling dummy indices, which is usually achieved by defining a canonical form for tensor product expressions. However, the problem of finding a canonical form is nontrivial, and can be potentially of exponential cost in the number of indices. In this work, we provided a general solution to this tensor canonicalization problem.




 An $\mathit{ab\ initio}$ theory is devised for the quantum dynamics of molecules undergoing (multiple) ionization in ultrafast and intense light. Specifically, the intertwined problem of photoionization, radiative, and electronic transitions in the course of dissociation is addressed which arises, e.g., when molecules are exposed to XUV light or x rays from free electron lasers or attosecond light sources, but the approach is equally useful in optical strong-field physics. The coherent interaction of the molecule with the light in a specific charge state is also treated. I set out from an abstract formulation in terms of the quantum optical notion of system-reservoir interaction using a master equation in Lindblad form and analyze its short-time approximation. First, I express it in a direct sum rigged Hilbert space for an efficient solution with numerical methods for systems of differential equations. Second, I derive a treatment via quantum Monte Carlo wave packet (MCWP) propagation. The formalism is concretized to diatomic molecules in Born-Oppenheimer approximation whereby molecular rotation is disregarded. The numerical integration of the master equation is carried out with a suitably factored density matrix that exploits the locality of the Hamiltonian and the Lindblad superoperator with respect to the internuclear distance. The formulation of the MCWP for molecules requires a thorough analysis of the quantum jump process; namely, the dependence on the continuous distance renders a straight wave packet promotion useless and, instead, a projected outer product needs to be employed involving an integrated quantum jump operator.
An $\mathit{ab\ initio}$ theory is devised for the quantum dynamics of molecules undergoing (multiple) ionization in ultrafast and intense light. Specifically, the intertwined problem of photoionization, radiative, and electronic transitions in the course of dissociation is addressed which arises, e.g., when molecules are exposed to XUV light or x rays from free electron lasers or attosecond light sources, but the approach is equally useful in optical strong-field physics. The coherent interaction of the molecule with the light in a specific charge state is also treated. I set out from an abstract formulation in terms of the quantum optical notion of system-reservoir interaction using a master equation in Lindblad form and analyze its short-time approximation. First, I express it in a direct sum rigged Hilbert space for an efficient solution with numerical methods for systems of differential equations. Second, I derive a treatment via quantum Monte Carlo wave packet (MCWP) propagation. The formalism is concretized to diatomic molecules in Born-Oppenheimer approximation whereby molecular rotation is disregarded. The numerical integration of the master equation is carried out with a suitably factored density matrix that exploits the locality of the Hamiltonian and the Lindblad superoperator with respect to the internuclear distance. The formulation of the MCWP for molecules requires a thorough analysis of the quantum jump process; namely, the dependence on the continuous distance renders a straight wave packet promotion useless and, instead, a projected outer product needs to be employed involving an integrated quantum jump operator.




 To assist the design of novel, highly efficient molecular junctions, a deep understanding of the precise charge transport mechanisms through these devices is of prime importance. In the present contribution, we describe a procedure to investigate spatially-resolved electron transport through a nanojunction from first principles, at the example of a nitro-substituted oligo-(phenylene ethynylene) covalently bound to graphene nanoribbon leads. Recently, we demonstrated that the conductivity of this single-molecule-graphene-nanoribbon junction can be switched quantitatively and reversibly upon application of a static electric field in a top gate position, in the spirit of a traditional field effect transistor [J. Phys. Chem. C, 2016, 120, 28808-28819]. The propensity of the central oligomer unit to align with the external field was found to induce a damped rotational motion and to cause an interruption of the conjugated $\pi$-system, thereby drastically reducing the conductance through the nanojunction. In the current work, we use the driven Liouville-von-Neumann (DLvN) approach for time-dependent electronic transport calculations to simulate the electronic current dynamics under time-dependent potential biases for the two logical states of the nanojunction. Our quantum dynamical simulations rely on a novel localization procedure using an orthonormal set of molecular orbitals obtained from a standard density functional theory calculation to generate a localized representation for the different parts of the molecular junction. The transparent DLvN formalism allows us to directly access the density matrix and to reconstruct the time-dependent electronic current density, unraveling unique mechanistic details of the electron transport.
To assist the design of novel, highly efficient molecular junctions, a deep understanding of the precise charge transport mechanisms through these devices is of prime importance. In the present contribution, we describe a procedure to investigate spatially-resolved electron transport through a nanojunction from first principles, at the example of a nitro-substituted oligo-(phenylene ethynylene) covalently bound to graphene nanoribbon leads. Recently, we demonstrated that the conductivity of this single-molecule-graphene-nanoribbon junction can be switched quantitatively and reversibly upon application of a static electric field in a top gate position, in the spirit of a traditional field effect transistor [J. Phys. Chem. C, 2016, 120, 28808-28819]. The propensity of the central oligomer unit to align with the external field was found to induce a damped rotational motion and to cause an interruption of the conjugated $\pi$-system, thereby drastically reducing the conductance through the nanojunction. In the current work, we use the driven Liouville-von-Neumann (DLvN) approach for time-dependent electronic transport calculations to simulate the electronic current dynamics under time-dependent potential biases for the two logical states of the nanojunction. Our quantum dynamical simulations rely on a novel localization procedure using an orthonormal set of molecular orbitals obtained from a standard density functional theory calculation to generate a localized representation for the different parts of the molecular junction. The transparent DLvN formalism allows us to directly access the density matrix and to reconstruct the time-dependent electronic current density, unraveling unique mechanistic details of the electron transport.


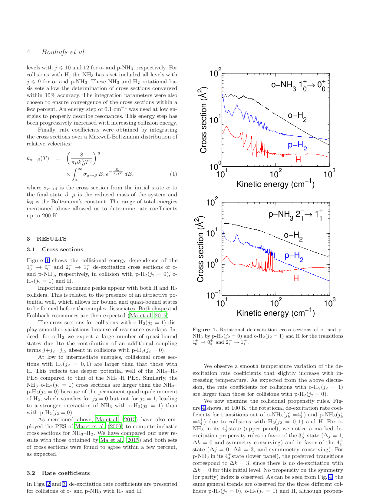
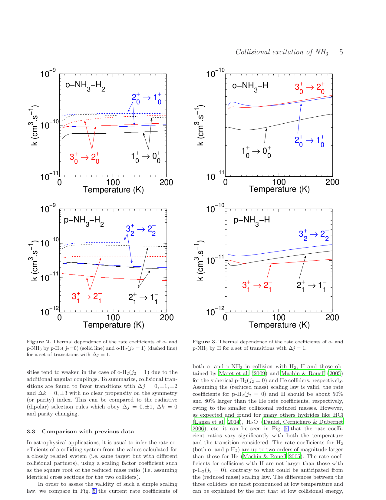
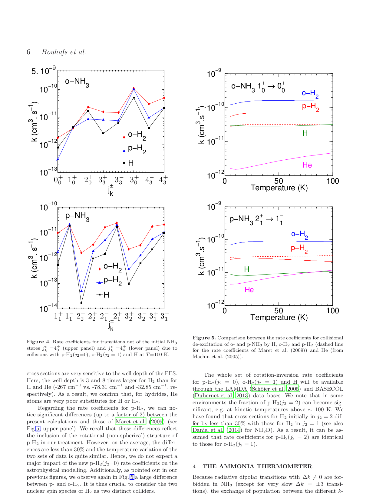 We report extensive theoretical calculations on the rotation-inversion excitation of interstellar ammonia (NH3) due to collisions with atomic and molecular hydrogen (both para- and ortho-H2). Close-coupling calculations are performed for total energies in the range 1-2000 cm-1 and rotational cross sections are obtained for all transitions among the lowest 17 and 34 rotation-inversion levels of ortho- and para-NH3, respectively. Rate coefficients are deduced for kinetic temperatures up to 200 K. Propensity rules for the three colliding partners are discussed and we also compare the new results to previous calculations for the spherically symmetrical He and para-H2 projectiles. Significant differences are found between the different sets of calculations. Finally, we test the impact of the new rate coefficients on the calibration of the ammonia thermometer. We find that the calibration curve is only weakly sensitive to the colliding partner and we confirm that the ammonia thermometer is robust.
We report extensive theoretical calculations on the rotation-inversion excitation of interstellar ammonia (NH3) due to collisions with atomic and molecular hydrogen (both para- and ortho-H2). Close-coupling calculations are performed for total energies in the range 1-2000 cm-1 and rotational cross sections are obtained for all transitions among the lowest 17 and 34 rotation-inversion levels of ortho- and para-NH3, respectively. Rate coefficients are deduced for kinetic temperatures up to 200 K. Propensity rules for the three colliding partners are discussed and we also compare the new results to previous calculations for the spherically symmetrical He and para-H2 projectiles. Significant differences are found between the different sets of calculations. Finally, we test the impact of the new rate coefficients on the calibration of the ammonia thermometer. We find that the calibration curve is only weakly sensitive to the colliding partner and we confirm that the ammonia thermometer is robust.

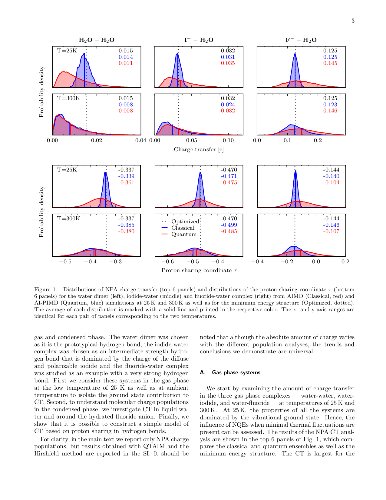
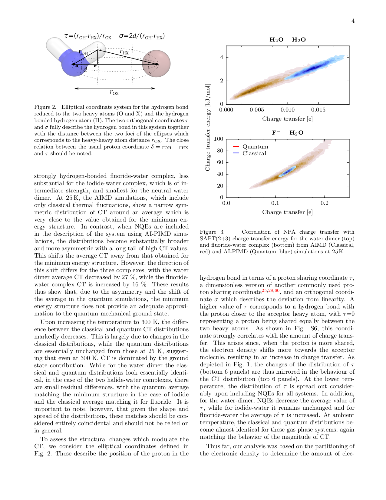
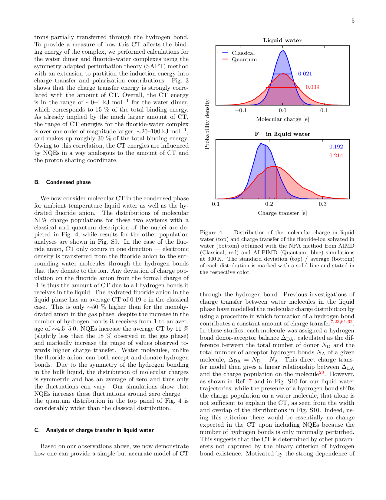
 Upon hydrogen bond formation, electronic charge density is transferred between the donor and acceptor, impacting processes ranging from hydration to spectroscopy. Here we use ab initio path integral simulations to elucidate the role of nuclear quantum effects in determining the charge transfer in a range of hydrogen bonded species in the gas and liquid phase. We show that the quantization of the nuclei gives rise to large changes in the magnitude of the charge transfer as well as its temperature dependence. We then explain how a single geometric parameter determines the charge transfer through the hydrogen bond. These results thus demonstrate that nuclear quantum effects are vital for the accurate description of charge transfer and offer a physically transparent way to understand how hydrogen bonding gives rise to it.
Upon hydrogen bond formation, electronic charge density is transferred between the donor and acceptor, impacting processes ranging from hydration to spectroscopy. Here we use ab initio path integral simulations to elucidate the role of nuclear quantum effects in determining the charge transfer in a range of hydrogen bonded species in the gas and liquid phase. We show that the quantization of the nuclei gives rise to large changes in the magnitude of the charge transfer as well as its temperature dependence. We then explain how a single geometric parameter determines the charge transfer through the hydrogen bond. These results thus demonstrate that nuclear quantum effects are vital for the accurate description of charge transfer and offer a physically transparent way to understand how hydrogen bonding gives rise to it.