-
A quantitative understanding of organism-level behavior requires predictive
models that can capture the richness of behavioral phenotypes, yet are simple
enough to connect with underlying mechanistic processes. Here we investigate
the motile behavior of nematodes at the level of their translational motion on
surfaces driven by undulatory propulsion. We broadly sample the nematode
behavioral repertoire by measuring motile trajectories of the canonical lab
strain $C. elegans$ N2 as well as wild strains and distant species. We focus on
trajectory dynamics over timescales spanning the transition from ballistic
(straight) to diffusive (random) movement and find that salient features of the
motility statistics are captured by a random walk model with independent
dynamics in the speed, bearing and reversal events. We show that the model
parameters vary among species in a correlated, low-dimensional manner
suggestive of a common mode of behavioral control and a trade-off between
exploration and exploitation. The distribution of phenotypes along this primary
mode of variation reveals that not only the mean but also the variance varies
considerably across strains, suggesting that these nematode lineages employ
contrasting ``bet-hedging'' strategies for foraging.
-
This paper introduces a theoretical framework for the analysis and control of
the stochastic susceptible-infected-removed (SIR) spreading process over a
network of heterogeneous agents. In our analysis, we analyze the exact
networked Markov process describing the SIR model, without resorting to
mean-field approximations, and introduce a convex optimization framework to
find an efficient allocation of resources to contain the expected number of
accumulated infections over time. Numerical simulations are presented to
illustrate the effectiveness of the obtained results.
-
Even this saying itself is a variant of a similar statement attributed to
Bernard of Chartres in the 12th Century, and inspired the title for a book by
Steven Hawking and an album by Oasis. Creative ideas beget other creative ideas
and, as a result, modifications accumulate, and we see an overall increase in
the complexity of cultural novelty over time, a phenomenon sometimes referred
to as the ratchet effect (Tomasello, Kruger, & Ratner, 1993). Although we may
never meet the people or objects that creatively influence us, by assimilating
what we encounter around us and bringing to bear our own insights and
perspectives, we all contribute in our own way, however small, to a second
evolutionary process -- the evolution of culture. This chapter explores how we
can better understand culture by understanding the creative processes that fuel
it, and better understand creativity by examining it from its cultural context.
First, we look at some theoretical frameworks for how culture evolves and what
these frameworks imply for the role of creativity. Then we will see how
questions about the relationship between creativity and cultural evolution have
been addressed using an agent-based model. We will also discuss studies of how
creative outputs are influenced, in perhaps unexpected ways, by other ideas and
individuals, and how individual creative styles "peek through" cultural outputs
in different domains.
-
This chapter takes as its departure point a neural level theory of insight
that arose from studies of the sparse, distributed, content-addressable
architecture of associative memory. It is argued that convergent thought is
most fruitfully characterized in terms of, not the generation of a single
correct solution (as it is conventionally construed), but using concepts in
their most compact form by activating neural cell assemblies that respond to
their most typical properties. This allows them to be deployed in a
conventional manner such that effort is reserved for exploring causal
relationships. Conversely, it is argued that divergent thought is most
fruitfully characterized in terms of, not the generation of multiple solutions
(as it is conventionally construed), but using concepts in a form that is,
albeit expanded, constrained by the situation, by activating neural cell
assemblies that respond to context-specific atypical properties. This allows
them to be deployed in a manner that is conducive to exploring unconventional
yet potentially relevant associations, and unearthing potentially useful
relationships of correlation. Thus, divergent thought can involve as few as one
idea. This proposal is compatible with widespread beliefs that (1) most
creative tasks require not many solutions but one, yet entail both divergent
and convergent thinking, and (2) not all problems with multiple solutions
require creative thinking, and conversely, some problems with single solution
do require creative thought. The chapter discusses how the ability to shift
between convergent and divergent modes of thought may have evolved, and it
concludes with educational and vocational implications.
-
Although viral spreading processes taking place in networks are often
analyzed using Markovian models in which both the transmission and the recovery
times follow exponential distributions, empirical studies show that, in many
real scenarios, the distribution of these times are not necessarily
exponential. To overcome this limitation, we first introduce a generalized
susceptible-infected-susceptible (SIS) spreading model that allows transmission
and recovery times to follow phase-type distributions. In this context, we
derive a lower bound on the exponential decay rate towards the infection-free
equilibrium of the spreading model without relying on mean-field
approximations. Based on our results, we illustrate how the particular shape of
the transmission/recovery distribution influences the exponential rate of
convergence towards the equilibrium.
-
Identifying the physical basis of heterosis (or hybrid vigor) has remained
elusive despite over a hundred years of research on the subject. The three main
theories of heterosis are dominance theory, overdominance theory, and epistasis
theory. Kacser and Burns (1981) identified the molecular basis of dominance,
which has greatly enhanced our understanding of its importance to heterosis.
This paper aims to explain how overdominance, and some features of epistasis,
can similarly emerge from the molecular dynamics of proteins. Possessing
multiple alleles at a gene locus results in the synthesis of different
allozymes at reduced concentrations. This in turn reduces the rate at which
each allozyme forms soluble oligomers, which are toxic and must be degraded,
because allozymes co-aggregate at low efficiencies. The model developed in this
paper will be used to explain how heterozygosity can impact the metabolic
efficiency of an organism. It can also explain why the viabilities of some
inbred lines seem to decline rapidly at high inbreeding coefficients (F > 0.5),
which may provide a physical basis for truncation selection for heterozygosity.
Finally, the model has implications for the ploidy level of organisms. It can
explain why polyploids are frequently found in environments where severe
physical stresses promote the formation of soluble oligomers. The model can
also explain why complex organisms, which need to synthesize aggregation-prone
proteins that contain intrinsically unstructured regions (IURs) and multiple
domains because they facilitate complex protein interaction networks (PINs),
tend to be diploid while haploidy tends to be restricted to relatively simple
organisms.
-
In this note, we characterize the embeddability of generic Kimura 3ST Markov
matrices in terms of their eigenvalues. As a consequence, we are able to
compute the volume of such matrices relative to the volume of all Markov
matrices within the model. We also provide examples showing that, in general,
mutation rates are not identifiable from substitution probabilities. These
examples also illustrate that symmetries between mutation probabilities do not
necessarily arise from symmetries between the corresponding mutation rates.
-
Meaningful laws of nature must be independent of the units employed to
measure the variables. The principle of similitude (Rayleigh 1915) or
dimensional homogeneity, states that only commensurable quantities (ones having
the same dimension) may be compared, therefore, meaningful laws of nature must
be homogeneous equations in their various units of measurement, a result which
was formalized in the $\rm \Pi$ theorem (Vaschy 1892; Buckingham 1914).
However, most relations in allometry do not satisfy this basic requirement,
including the `3/4 Law' (Kleiber 1932) that relates the basal metabolic rate
and body mass, which it is sometimes claimed to be the most fundamental
biological rate (Brown et al. 2004) and the closest to a law in life sciences
(West \& Brown 2004). Using the $\rm \Pi$ theorem, here we show that it is
possible to construct a unique homogeneous equation for the metabolic rates, in
agreement with data in the literature. We find that the variations in the
dependence of the metabolic rates on body mass are secondary, coming from
variations in the allometric dependence of the heart frequencies. This includes
not only different classes of animals (mammals, birds, invertebrates) but also
different exercise conditions (basal and maximal). Our results demonstrate that
most of the differences found in the allometric exponents (White et al. 2007)
are due to compare incommensurable quantities and that our dimensionally
homogenous formula, unify these differences into a single formulation. We
discuss the ecological implications of this new formulation in the context of
the Malthusian's, Fenchel's and the total energy consumed in a lifespan
relations.
-
In addition to their unusually long life cycle, periodical cicadas, {\it
Magicicada} spp., provide an exceptional example of spatially synchronized life
stage phenology in nature. Within regions ("broods") spanning 50,000 to 500,000
km$^2$, adults emerge synchronously every 13 or 17 years. While satiation of
avian predators is believed to be a key component of the ability of these
populations to reach high densities, it is not clear why populations at a
single location remain entirely synchronized. We develop nonlinear Leslie
matrix-type models of periodical cicadas that include predation-driven Allee
effects and competition in addition to reproduction and survival. Using both
analytical and numerical techniques, we demonstrate the observed presence of a
single brood critically depends on the relationship between fecundity,
competition, and predation. We analyze the single-brood, two-brood and
all-brood equilibria in the large life-span limit using a tractable hybrid
approximation to the Leslie matrix model with continuous time competition in
between discrete reproduction events. Within the hybrid model we prove that the
single-brood equilibrium is the only stable equilibrium. This hybrid model
allows us to quantitatively predict population sizes and the range of
parameters for which the stable single-brood and unstable two-brood and
all-brood equilibria exist. The hybrid model yields a good approximation to the
numerical results for the Leslie matrix model for the biologically relevant
case of a 17-year lifespan.
-
Identifying undocumented or potential future interactions among species is a
challenge facing modern ecologists. Recent link prediction methods rely on
trait data, however large species interaction databases are typically sparse
and covariates are limited to only a fraction of species. On the other hand,
evolutionary relationships, encoded as phylogenetic trees, can act as proxies
for underlying traits and historical patterns of parasite sharing among hosts.
We show that using a network-based conditional model, phylogenetic information
provides strong predictive power in a recently published global database of
host-parasite interactions. By scaling the phylogeny using an evolutionary
model, our method allows for biological interpretation often missing from
latent variable models. To further improve on the phylogeny-only model, we
combine a hierarchical Bayesian latent score framework for bipartite graphs
that accounts for the number of interactions per species with the host
dependence informed by phylogeny. Combining the two information sources yields
significant improvement in predictive accuracy over each of the submodels
alone. As many interaction networks are constructed from presence-only data, we
extend the model by integrating a correction mechanism for missing
interactions, which proves valuable in reducing uncertainty in unobserved
interactions.
-
Many mathematical models of evolution assume that all individuals experience
the same environment. Here, we study the Moran process in heterogeneous
environments. The population is of finite size with two competing types, which
are exposed to a fixed number of environmental conditions. Reproductive rate is
determined by both the type and the environment. We first calculate the
condition for selection to favor the mutant relative to the resident wild type.
In large populations, the mutant is favored if and only if the mutant's spatial
average reproductive rate exceeds that of the resident. But environmental
heterogeneity elucidates an interesting asymmetry between the mutant and the
resident. Specifically, mutant heterogeneity suppresses its fixation
probability; if this heterogeneity is strong enough, it can even completely
offset the effects of selection (including in large populations). In contrast,
resident heterogeneity has no effect on a mutant's fixation probability in
large populations and can amplify it in small populations.
-
In large clonal populations, several clones generally compete which results
in complex evolutionary and ecological dynamics: experiments show successive
selective sweeps of favorable mutations as well as long-term coexistence of
multiple clonal strains. The mechanisms underlying either coexistence or
fixation of several competing strains have rarely been studied altogether.
Conditions for coexistence have mostly been studied by population and community
ecology, while rates of invasion and fixation have mostly been studied by
population genetics. In order to provide a global understanding of the
complexity of the dynamics observed in large clonal populations, we develop a
stochastic model where three clones compete. Competitive interactions can be
intransitive and we suppose that strains enter the population via mutations or
rare immigrations. We first describe all possible final states of the
population, including stable coexistence of two or three strains, or the
fixation of a single strain. Second, we give estimate of the invasion and
fixation times of a favorable mutant (or immigrant) entering the population in
a single copy. We show that invasion and fixation can be slower or faster when
considering complex competitive interactions. Third, we explore the parameter
space assuming prior distributions of reproduction, death and competitive rates
and we estimate the likelihood of the possible dynamics. We show that when
mutations can affect competitive interactions, even slightly, stable
coexistence is likely. We discuss our results in the context of the
evolutionary dynamics of large clonal populations.
-
Transcription factors (TFs) exert their regulatory action by binding to DNA
with specific sequence preferences. However, different TFs can partially share
their binding sequences due to their common evolutionary origin. This
`redundancy' of binding defines a way of organizing TFs in `motif families' by
grouping TFs with similar binding preferences. Since these ultimately define
the TF target genes, the motif family organization entails information about
the structure of transcriptional regulation as it has been shaped by evolution.
Focusing on the human TF repertoire, we show that a one-parameter evolutionary
model of the Birth-Death-Innovation type can explain the TF empirical
ripartition in motif families, and allows to highlight the relevant
evolutionary forces at the origin of this organization. Moreover, the model
allows to pinpoint few deviations from the neutral scenario it assumes: three
over-expanded families (including HOX and FOX genes), a set of `singleton' TFs
for which duplication seems to be selected against, and a higher-than-average
rate of diversification of the binding preferences of TFs with a Zinc Finger
DNA binding domain. Finally, a comparison of the TF motif family organization
in different eukaryotic species suggests an increase of redundancy of binding
with organism complexity.
-
Recent technological changes have increased connectivity between individuals
around the world leading to higher frequency interactions between members of
communities that would be otherwise distant and disconnected. This paper
examines a model of opinion dynamics in interacting communities and studies how
increasing interaction frequency affects the ability for communities to retain
distinct identities versus falling into consensus or polarized states in which
community identity is lost. We also study the effect (if any) of opinion noise
related to a tendency for individuals to assert their individuality in
homogenous populations. Our work builds on a model we developed previously [11]
where the dynamics of opinion change is based on individual interactions that
seek to minimize some energy potential based on the differences between
opinions across the population.
-
In this paper we propose a method to study a general vector-hosts
mathematical model in order to explain how the changes in biodiversity could
influence the dynamics of vector-borne diseases. We find that under the
assumption of frequency-dependent transmission, i.e. the assumption that the
number of contacts are diluted by the total population of hosts, the presence
of a competent host is a necessary condition for the existence of an endemic
state. In addition, we obtain that in the case of an endemic disease with a
unique competent and resilient host, an increase in its density amplifies the
disease.
-
We consider a class of birth-and-death processes describing a population made
of $d$ sub-populations of different types which interact with one another. The
state space is $\mathbb{Z}_+^d$ (unbounded). We assume that the population goes
almost surely to extinction, so that the unique stationary distribution is the
Dirac measure at the origin. These processes are parametrized by a scaling
parameter $K$ which can be thought as the order of magnitude of the total size
of the population at time $0$. For any fixed finite time span, it is well-known
that such processes, when renormalized by $K$, are close, in the limit
$K\to+\infty$, to the solutions of a certain differential equation in
$\mathbb{R}_+^d$ whose vector field is determined by the birth and death rates.
We consider the case where there is a unique attractive fixed point (off the
boundary of the positive orthant) for the vector field (while the origin is
repulsive). What is expected is that, for $K$ large, the process will stay in
the vicinity of the fixed point for a very long time before being absorbed at
the origin. To precisely describe this behavior, we prove the existence of a
quasi-stationary distribution (qsd). In fact, we establish a bound for the
total variation distance between the process conditioned to non-extinction
before time $t$ and the qsd. This bound is exponentially small in $t$, for
$t\gg \log K$. As a by-product, we obtain an estimate for the mean time to
extinction in the qsd. We also quantify how close is the law of the process
(not conditioned to non-extinction) either to the Dirac measure at the origin
or to the qsd, for times much larger than $\log K$ and much smaller than the
mean time to extinction, which is exponentially large as a function of $K$. Let
us stress that we are interested in what happens for finite $K$. We obtain
results much beyond what large deviation techniques could provide.
-
An elementary biostatistical theory based on a selectivity-variability
principle is proposed to address a question raised by Charles Darwin, namely,
how one sex of a sexually dimorphic species might tend to evolve with greater
variability than the other sex. Briefly, the theory says that if one sex is
relatively selective then from one generation to the next, more variable
subpopulations of the opposite sex will generally tend to prevail over those
with lesser variability. Moreover, the perhaps less intuitive converse also
holds: if a sex is relatively non-selective, then less variable subpopulations
of the opposite sex will prevail over those with greater variability. This
theory requires certain regularity conditions on the distributions, but makes
no assumptions about differences in means between the sexes, nor does it
presume that one sex is selective and the other non-selective. Two mathematical
models of the selectivity-variability principle are presented: a discrete-time
one-step probabilistic model of short-term behavior with an example using
normally distributed perceived fitness values; and a continuous-time
deterministic model for the long-term asymptotic behavior of the expected sizes
of the subpopulations with an example using exponentially distributed fitness
levels.
-
The Moran model with recombination is considered, which describes the
evolution of the genetic composition of a population under recombination and
resampling. There are $n$ sites (or loci), a finite number of letters (or
alleles) at every site, and we do not make any scaling assumptions. In
particular, we do not assume a diffusion limit. We consider the following
marginal ancestral recombination process. Let $S = \{1,...,n\}$ and $\mathcal
A=\{A_1, ..., A_m\}$ be a partition of $S$. We concentrate on the joint
probability of the letters at the sites in $A_1$ in individual $1$, $...$, and
at the sites in $A_m$ in individual $m$, where the individuals are sampled from
the current population without replacement. Following the ancestry of these
sites backwards in time yields a process on the set of partitions of $S$,
which, in the diffusion limit, turns into a marginalised version of the
$n$-locus ancestral recombination graph. With the help of an
inclusion-exclusion principle, we show that the type distribution corresponding
to a given partition may be represented in a systematic way, with the help of
so-called recombinators and sampling functions. The same is true of correlation
functions (known as linkage disequilibria in genetics) of all orders.
We prove that the partitioning process (backward in time) is dual to the
Moran population process (forward in time), where the sampling function plays
the role of the duality function. This sheds new light on the work of
Bobrowski, Wojdyla, and Kimmel (2010). The result also leads to a closed system
of ordinary differential equations for the expectations of the sampling
functions, which can be translated into expected type distributions and
expected linkage disequilibria.
-
The distributions of the times to the first common ancestor t_mrca is
numerically studied for an ecological population model, the extended Moran
model. This model has a fixed population size N. The number of descendants is
drawn from a beta distribution Beta(alpha, 2-alpha) for various choices of
alpha. This includes also the classical Moran model (alpha->0) as well as the
uniform distribution (alpha=1). Using a statistical mechanics-based
large-deviation approach, the distributions can be studied over an extended
range of the support, down to probabilities like 10^{-70}, which allowed us to
study the change of the tails of the distribution when varying the value of
alpha in [0,2]. We find exponential distributions p(t_mrca)~ delta^{t_mrca} in
all cases, with systematically varying values for the base delta. Only for the
cases alpha=0 and alpha=1, analytical results are known, i.e.,
delta=\exp(-2/N^2) and delta=2/3, respectively. We recover these values,
confirming the validity of our approach. Finally, we also study the
correlations between t_mrca and the number of descendants.
-
Topological phylogenetic trees can be assigned edge weights in several
natural ways, highlighting different aspects of the tree. Here the rooted
triple and quartet metrizations are introduced, and applied to formulate novel
fast methods of inferring large trees from rooted triple and quartet data.
These methods can be applied in new statistically consistent procedures for
inference of a species tree from gene trees under the multispecies coalescent
model.
-
Infectious disease outbreaks recapitulate biology: they emerge from the
multi-level interaction of hosts, pathogens, and their shared environment. As a
result, predicting when, where, and how far diseases will spread requires a
complex systems approach to modeling. Recent studies have demonstrated that
predicting different components of outbreaks--e.g., the expected number of
cases, pace and tempo of cases needing treatment, demand for prophylactic
equipment, importation probability etc.--is feasible. Therefore, advancing both
the science and practice of disease forecasting now requires testing for the
presence of fundamental limits to outbreak prediction. To investigate the
question of outbreak prediction, we study the information theoretic limits to
forecasting across a broad set of infectious diseases using permutation entropy
as a model independent measure of predictability. Studying the predictability
of a diverse collection of historical outbreaks--including, chlamydia, dengue,
gonorrhea, hepatitis A, influenza, measles, mumps, polio, and whooping
cough--we identify a fundamental entropy barrier for infectious disease time
series forecasting. However, we find that for most diseases this barrier to
prediction is often well beyond the time scale of single outbreaks. We also
find that the forecast horizon varies by disease and demonstrate that both
shifting model structures and social network heterogeneity are the most likely
mechanisms for the observed differences across contagions. Our results
highlight the importance of moving beyond time series forecasting, by embracing
dynamic modeling approaches, and suggest challenges for performing model
selection across long time series. We further anticipate that our findings will
contribute to the rapidly growing field of epidemiological forecasting and may
relate more broadly to the predictability of complex adaptive systems.
-
The stochastic dynamics of biochemical networks are usually modelled with the
chemical master equation (CME). The stationary distributions of CMEs are seldom
solvable analytically, and numerical methods typically produce estimates with
uncontrolled errors. Here, we introduce mathematical programming approaches
that yield approximations of these distributions with computable error bounds
which enable the verification of their accuracy. First, we use semidefinite
programming to compute increasingly tighter upper and lower bounds on the
moments of the stationary distributions for networks with rational
propensities. Second, we use these moment bounds to formulate linear programs
that yield convergent upper and lower bounds on the stationary distributions
themselves, their marginals and stationary averages. The bounds obtained also
provide a computational test for the uniqueness of the distribution. In the
unique case, the bounds form an approximation of the stationary distribution
with a computable bound on its error. In the non-unique case, our approach
yields converging approximations of the ergodic distributions. We illustrate
our methodology through several biochemical examples taken from the literature:
Schl\"ogl's model for a chemical bifurcation, a two-dimensional toggle switch,
a model for bursty gene expression, and a dimerisation model with multiple
stationary distributions.
-
The spread of invasive species can have far reaching environmental and
ecological consequences. Understanding invasion spread patterns and the
underlying process driving invasions are key to predicting and managing
invasions. We combine a set of statistical methods in a novel way to
characterize local spread properties and demonstrate their application using
simulated and historical data on invasive insects. Our method uses a Gaussian
process fit to the surface of waiting times to invasion in order to
characterize the vector field of spread. Using this method we estimate with
statistical uncertainties the speed and direction of spread at each location.
Simulations from a stratified diffusion model verify the accuracy of our
method. We show how we may link local rates of spread to environmental
covariates for two case studies: the spread of the gypsy moth (Lymantria
dispar), and hemlock wolly adelgid (Adelges tsugae) in North America. We
provide an R-package that automates the calculations for any spatially
referenced waiting time data.
-
One of the main aims of phylogenetics is to reconstruct the \enquote{Tree of
Life}. In this respect, different methods and criteria are used to analyze DNA
sequences of different species and to compare them in order to derive the
evolutionary relationships of these species. Maximum Parsimony is one such
criterion for tree reconstruction and, it is the one which we will use in this
paper. However, it is well-known that tree reconstruction methods can lead to
wrong relationship estimates. One typical problem of Maximum Parsimony is long
branch attraction, which can lead to statistical inconsistency. In this work,
we will consider a blockwise approach to alignment analysis, namely so-called
$k$-tuple analyses. For four taxa it has already been shown that
$k$-tuple-based analyses are statistically inconsistent if and only if the
standard character-based (site-based) analyses are statistically inconsistent.
So, in the four-taxon case, going from individual sites to $k$-tuples does not
lead to any improvement. However, real biological analyses often consider more
than only four taxa. Therefore, we analyze the case of five taxa for $2$- and
$3$-tuple-site data and consider alphabets with two and four elements. We show
that the equivalence of single-site data and $k$-tuple-site data then no longer
holds. Even so, we can show that Maximum Parsimony is statistically
inconsistent for $k$-tuple site data and five taxa.
-
We investigate the global dynamics of a general Kermack-McKendrick-type
epidemic model formulated in terms of a system of renewal equations.
Specifically, we consider a renewal model for which both the force of infection
and the infected removal rates are arbitrary functions of the infection age,
$\tau$, and use the direct Lyapunov method to establish the global asymptotic
stability of the equilibrium solutions. In particular, we show that the basic
reproduction number, $R_0$, represents a sharp threshold parameter such that
for $R_0\leq 1$, the infection-free equilibrium is globally asymptotically
stable; whereas the endemic equilibrium becomes globally asymptotically stable
when $R_0 > 1$, i.e. when it exists.





 A quantitative understanding of organism-level behavior requires predictive models that can capture the richness of behavioral phenotypes, yet are simple enough to connect with underlying mechanistic processes. Here we investigate the motile behavior of nematodes at the level of their translational motion on surfaces driven by undulatory propulsion. We broadly sample the nematode behavioral repertoire by measuring motile trajectories of the canonical lab strain $C. elegans$ N2 as well as wild strains and distant species. We focus on trajectory dynamics over timescales spanning the transition from ballistic (straight) to diffusive (random) movement and find that salient features of the motility statistics are captured by a random walk model with independent dynamics in the speed, bearing and reversal events. We show that the model parameters vary among species in a correlated, low-dimensional manner suggestive of a common mode of behavioral control and a trade-off between exploration and exploitation. The distribution of phenotypes along this primary mode of variation reveals that not only the mean but also the variance varies considerably across strains, suggesting that these nematode lineages employ contrasting ``bet-hedging'' strategies for foraging.
A quantitative understanding of organism-level behavior requires predictive models that can capture the richness of behavioral phenotypes, yet are simple enough to connect with underlying mechanistic processes. Here we investigate the motile behavior of nematodes at the level of their translational motion on surfaces driven by undulatory propulsion. We broadly sample the nematode behavioral repertoire by measuring motile trajectories of the canonical lab strain $C. elegans$ N2 as well as wild strains and distant species. We focus on trajectory dynamics over timescales spanning the transition from ballistic (straight) to diffusive (random) movement and find that salient features of the motility statistics are captured by a random walk model with independent dynamics in the speed, bearing and reversal events. We show that the model parameters vary among species in a correlated, low-dimensional manner suggestive of a common mode of behavioral control and a trade-off between exploration and exploitation. The distribution of phenotypes along this primary mode of variation reveals that not only the mean but also the variance varies considerably across strains, suggesting that these nematode lineages employ contrasting ``bet-hedging'' strategies for foraging.



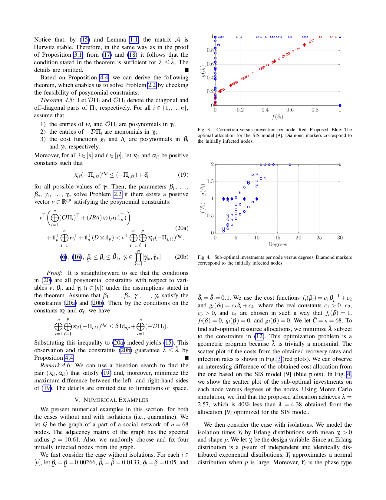
 This paper introduces a theoretical framework for the analysis and control of the stochastic susceptible-infected-removed (SIR) spreading process over a network of heterogeneous agents. In our analysis, we analyze the exact networked Markov process describing the SIR model, without resorting to mean-field approximations, and introduce a convex optimization framework to find an efficient allocation of resources to contain the expected number of accumulated infections over time. Numerical simulations are presented to illustrate the effectiveness of the obtained results.
This paper introduces a theoretical framework for the analysis and control of the stochastic susceptible-infected-removed (SIR) spreading process over a network of heterogeneous agents. In our analysis, we analyze the exact networked Markov process describing the SIR model, without resorting to mean-field approximations, and introduce a convex optimization framework to find an efficient allocation of resources to contain the expected number of accumulated infections over time. Numerical simulations are presented to illustrate the effectiveness of the obtained results.




 Although viral spreading processes taking place in networks are often analyzed using Markovian models in which both the transmission and the recovery times follow exponential distributions, empirical studies show that, in many real scenarios, the distribution of these times are not necessarily exponential. To overcome this limitation, we first introduce a generalized susceptible-infected-susceptible (SIS) spreading model that allows transmission and recovery times to follow phase-type distributions. In this context, we derive a lower bound on the exponential decay rate towards the infection-free equilibrium of the spreading model without relying on mean-field approximations. Based on our results, we illustrate how the particular shape of the transmission/recovery distribution influences the exponential rate of convergence towards the equilibrium.
Although viral spreading processes taking place in networks are often analyzed using Markovian models in which both the transmission and the recovery times follow exponential distributions, empirical studies show that, in many real scenarios, the distribution of these times are not necessarily exponential. To overcome this limitation, we first introduce a generalized susceptible-infected-susceptible (SIS) spreading model that allows transmission and recovery times to follow phase-type distributions. In this context, we derive a lower bound on the exponential decay rate towards the infection-free equilibrium of the spreading model without relying on mean-field approximations. Based on our results, we illustrate how the particular shape of the transmission/recovery distribution influences the exponential rate of convergence towards the equilibrium.




 Identifying the physical basis of heterosis (or hybrid vigor) has remained elusive despite over a hundred years of research on the subject. The three main theories of heterosis are dominance theory, overdominance theory, and epistasis theory. Kacser and Burns (1981) identified the molecular basis of dominance, which has greatly enhanced our understanding of its importance to heterosis. This paper aims to explain how overdominance, and some features of epistasis, can similarly emerge from the molecular dynamics of proteins. Possessing multiple alleles at a gene locus results in the synthesis of different allozymes at reduced concentrations. This in turn reduces the rate at which each allozyme forms soluble oligomers, which are toxic and must be degraded, because allozymes co-aggregate at low efficiencies. The model developed in this paper will be used to explain how heterozygosity can impact the metabolic efficiency of an organism. It can also explain why the viabilities of some inbred lines seem to decline rapidly at high inbreeding coefficients (F > 0.5), which may provide a physical basis for truncation selection for heterozygosity. Finally, the model has implications for the ploidy level of organisms. It can explain why polyploids are frequently found in environments where severe physical stresses promote the formation of soluble oligomers. The model can also explain why complex organisms, which need to synthesize aggregation-prone proteins that contain intrinsically unstructured regions (IURs) and multiple domains because they facilitate complex protein interaction networks (PINs), tend to be diploid while haploidy tends to be restricted to relatively simple organisms.
Identifying the physical basis of heterosis (or hybrid vigor) has remained elusive despite over a hundred years of research on the subject. The three main theories of heterosis are dominance theory, overdominance theory, and epistasis theory. Kacser and Burns (1981) identified the molecular basis of dominance, which has greatly enhanced our understanding of its importance to heterosis. This paper aims to explain how overdominance, and some features of epistasis, can similarly emerge from the molecular dynamics of proteins. Possessing multiple alleles at a gene locus results in the synthesis of different allozymes at reduced concentrations. This in turn reduces the rate at which each allozyme forms soluble oligomers, which are toxic and must be degraded, because allozymes co-aggregate at low efficiencies. The model developed in this paper will be used to explain how heterozygosity can impact the metabolic efficiency of an organism. It can also explain why the viabilities of some inbred lines seem to decline rapidly at high inbreeding coefficients (F > 0.5), which may provide a physical basis for truncation selection for heterozygosity. Finally, the model has implications for the ploidy level of organisms. It can explain why polyploids are frequently found in environments where severe physical stresses promote the formation of soluble oligomers. The model can also explain why complex organisms, which need to synthesize aggregation-prone proteins that contain intrinsically unstructured regions (IURs) and multiple domains because they facilitate complex protein interaction networks (PINs), tend to be diploid while haploidy tends to be restricted to relatively simple organisms.




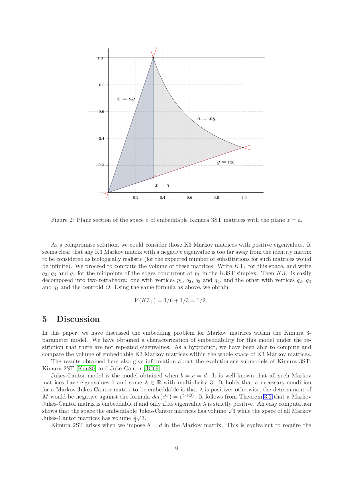 In this note, we characterize the embeddability of generic Kimura 3ST Markov matrices in terms of their eigenvalues. As a consequence, we are able to compute the volume of such matrices relative to the volume of all Markov matrices within the model. We also provide examples showing that, in general, mutation rates are not identifiable from substitution probabilities. These examples also illustrate that symmetries between mutation probabilities do not necessarily arise from symmetries between the corresponding mutation rates.
In this note, we characterize the embeddability of generic Kimura 3ST Markov matrices in terms of their eigenvalues. As a consequence, we are able to compute the volume of such matrices relative to the volume of all Markov matrices within the model. We also provide examples showing that, in general, mutation rates are not identifiable from substitution probabilities. These examples also illustrate that symmetries between mutation probabilities do not necessarily arise from symmetries between the corresponding mutation rates.




 Meaningful laws of nature must be independent of the units employed to measure the variables. The principle of similitude (Rayleigh 1915) or dimensional homogeneity, states that only commensurable quantities (ones having the same dimension) may be compared, therefore, meaningful laws of nature must be homogeneous equations in their various units of measurement, a result which was formalized in the $\rm \Pi$ theorem (Vaschy 1892; Buckingham 1914). However, most relations in allometry do not satisfy this basic requirement, including the `3/4 Law' (Kleiber 1932) that relates the basal metabolic rate and body mass, which it is sometimes claimed to be the most fundamental biological rate (Brown et al. 2004) and the closest to a law in life sciences (West \& Brown 2004). Using the $\rm \Pi$ theorem, here we show that it is possible to construct a unique homogeneous equation for the metabolic rates, in agreement with data in the literature. We find that the variations in the dependence of the metabolic rates on body mass are secondary, coming from variations in the allometric dependence of the heart frequencies. This includes not only different classes of animals (mammals, birds, invertebrates) but also different exercise conditions (basal and maximal). Our results demonstrate that most of the differences found in the allometric exponents (White et al. 2007) are due to compare incommensurable quantities and that our dimensionally homogenous formula, unify these differences into a single formulation. We discuss the ecological implications of this new formulation in the context of the Malthusian's, Fenchel's and the total energy consumed in a lifespan relations.
Meaningful laws of nature must be independent of the units employed to measure the variables. The principle of similitude (Rayleigh 1915) or dimensional homogeneity, states that only commensurable quantities (ones having the same dimension) may be compared, therefore, meaningful laws of nature must be homogeneous equations in their various units of measurement, a result which was formalized in the $\rm \Pi$ theorem (Vaschy 1892; Buckingham 1914). However, most relations in allometry do not satisfy this basic requirement, including the `3/4 Law' (Kleiber 1932) that relates the basal metabolic rate and body mass, which it is sometimes claimed to be the most fundamental biological rate (Brown et al. 2004) and the closest to a law in life sciences (West \& Brown 2004). Using the $\rm \Pi$ theorem, here we show that it is possible to construct a unique homogeneous equation for the metabolic rates, in agreement with data in the literature. We find that the variations in the dependence of the metabolic rates on body mass are secondary, coming from variations in the allometric dependence of the heart frequencies. This includes not only different classes of animals (mammals, birds, invertebrates) but also different exercise conditions (basal and maximal). Our results demonstrate that most of the differences found in the allometric exponents (White et al. 2007) are due to compare incommensurable quantities and that our dimensionally homogenous formula, unify these differences into a single formulation. We discuss the ecological implications of this new formulation in the context of the Malthusian's, Fenchel's and the total energy consumed in a lifespan relations.




 In addition to their unusually long life cycle, periodical cicadas, {\it Magicicada} spp., provide an exceptional example of spatially synchronized life stage phenology in nature. Within regions ("broods") spanning 50,000 to 500,000 km$^2$, adults emerge synchronously every 13 or 17 years. While satiation of avian predators is believed to be a key component of the ability of these populations to reach high densities, it is not clear why populations at a single location remain entirely synchronized. We develop nonlinear Leslie matrix-type models of periodical cicadas that include predation-driven Allee effects and competition in addition to reproduction and survival. Using both analytical and numerical techniques, we demonstrate the observed presence of a single brood critically depends on the relationship between fecundity, competition, and predation. We analyze the single-brood, two-brood and all-brood equilibria in the large life-span limit using a tractable hybrid approximation to the Leslie matrix model with continuous time competition in between discrete reproduction events. Within the hybrid model we prove that the single-brood equilibrium is the only stable equilibrium. This hybrid model allows us to quantitatively predict population sizes and the range of parameters for which the stable single-brood and unstable two-brood and all-brood equilibria exist. The hybrid model yields a good approximation to the numerical results for the Leslie matrix model for the biologically relevant case of a 17-year lifespan.
In addition to their unusually long life cycle, periodical cicadas, {\it Magicicada} spp., provide an exceptional example of spatially synchronized life stage phenology in nature. Within regions ("broods") spanning 50,000 to 500,000 km$^2$, adults emerge synchronously every 13 or 17 years. While satiation of avian predators is believed to be a key component of the ability of these populations to reach high densities, it is not clear why populations at a single location remain entirely synchronized. We develop nonlinear Leslie matrix-type models of periodical cicadas that include predation-driven Allee effects and competition in addition to reproduction and survival. Using both analytical and numerical techniques, we demonstrate the observed presence of a single brood critically depends on the relationship between fecundity, competition, and predation. We analyze the single-brood, two-brood and all-brood equilibria in the large life-span limit using a tractable hybrid approximation to the Leslie matrix model with continuous time competition in between discrete reproduction events. Within the hybrid model we prove that the single-brood equilibrium is the only stable equilibrium. This hybrid model allows us to quantitatively predict population sizes and the range of parameters for which the stable single-brood and unstable two-brood and all-brood equilibria exist. The hybrid model yields a good approximation to the numerical results for the Leslie matrix model for the biologically relevant case of a 17-year lifespan.



 Identifying undocumented or potential future interactions among species is a challenge facing modern ecologists. Recent link prediction methods rely on trait data, however large species interaction databases are typically sparse and covariates are limited to only a fraction of species. On the other hand, evolutionary relationships, encoded as phylogenetic trees, can act as proxies for underlying traits and historical patterns of parasite sharing among hosts. We show that using a network-based conditional model, phylogenetic information provides strong predictive power in a recently published global database of host-parasite interactions. By scaling the phylogeny using an evolutionary model, our method allows for biological interpretation often missing from latent variable models. To further improve on the phylogeny-only model, we combine a hierarchical Bayesian latent score framework for bipartite graphs that accounts for the number of interactions per species with the host dependence informed by phylogeny. Combining the two information sources yields significant improvement in predictive accuracy over each of the submodels alone. As many interaction networks are constructed from presence-only data, we extend the model by integrating a correction mechanism for missing interactions, which proves valuable in reducing uncertainty in unobserved interactions.
Identifying undocumented or potential future interactions among species is a challenge facing modern ecologists. Recent link prediction methods rely on trait data, however large species interaction databases are typically sparse and covariates are limited to only a fraction of species. On the other hand, evolutionary relationships, encoded as phylogenetic trees, can act as proxies for underlying traits and historical patterns of parasite sharing among hosts. We show that using a network-based conditional model, phylogenetic information provides strong predictive power in a recently published global database of host-parasite interactions. By scaling the phylogeny using an evolutionary model, our method allows for biological interpretation often missing from latent variable models. To further improve on the phylogeny-only model, we combine a hierarchical Bayesian latent score framework for bipartite graphs that accounts for the number of interactions per species with the host dependence informed by phylogeny. Combining the two information sources yields significant improvement in predictive accuracy over each of the submodels alone. As many interaction networks are constructed from presence-only data, we extend the model by integrating a correction mechanism for missing interactions, which proves valuable in reducing uncertainty in unobserved interactions.




 In large clonal populations, several clones generally compete which results in complex evolutionary and ecological dynamics: experiments show successive selective sweeps of favorable mutations as well as long-term coexistence of multiple clonal strains. The mechanisms underlying either coexistence or fixation of several competing strains have rarely been studied altogether. Conditions for coexistence have mostly been studied by population and community ecology, while rates of invasion and fixation have mostly been studied by population genetics. In order to provide a global understanding of the complexity of the dynamics observed in large clonal populations, we develop a stochastic model where three clones compete. Competitive interactions can be intransitive and we suppose that strains enter the population via mutations or rare immigrations. We first describe all possible final states of the population, including stable coexistence of two or three strains, or the fixation of a single strain. Second, we give estimate of the invasion and fixation times of a favorable mutant (or immigrant) entering the population in a single copy. We show that invasion and fixation can be slower or faster when considering complex competitive interactions. Third, we explore the parameter space assuming prior distributions of reproduction, death and competitive rates and we estimate the likelihood of the possible dynamics. We show that when mutations can affect competitive interactions, even slightly, stable coexistence is likely. We discuss our results in the context of the evolutionary dynamics of large clonal populations.
In large clonal populations, several clones generally compete which results in complex evolutionary and ecological dynamics: experiments show successive selective sweeps of favorable mutations as well as long-term coexistence of multiple clonal strains. The mechanisms underlying either coexistence or fixation of several competing strains have rarely been studied altogether. Conditions for coexistence have mostly been studied by population and community ecology, while rates of invasion and fixation have mostly been studied by population genetics. In order to provide a global understanding of the complexity of the dynamics observed in large clonal populations, we develop a stochastic model where three clones compete. Competitive interactions can be intransitive and we suppose that strains enter the population via mutations or rare immigrations. We first describe all possible final states of the population, including stable coexistence of two or three strains, or the fixation of a single strain. Second, we give estimate of the invasion and fixation times of a favorable mutant (or immigrant) entering the population in a single copy. We show that invasion and fixation can be slower or faster when considering complex competitive interactions. Third, we explore the parameter space assuming prior distributions of reproduction, death and competitive rates and we estimate the likelihood of the possible dynamics. We show that when mutations can affect competitive interactions, even slightly, stable coexistence is likely. We discuss our results in the context of the evolutionary dynamics of large clonal populations.


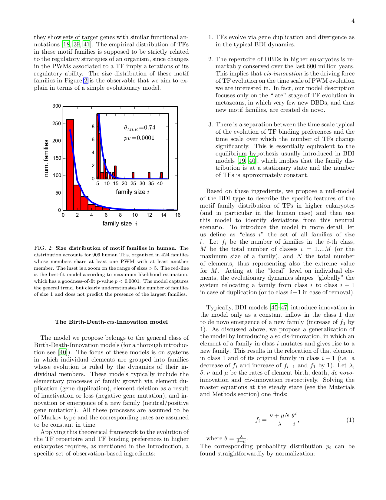

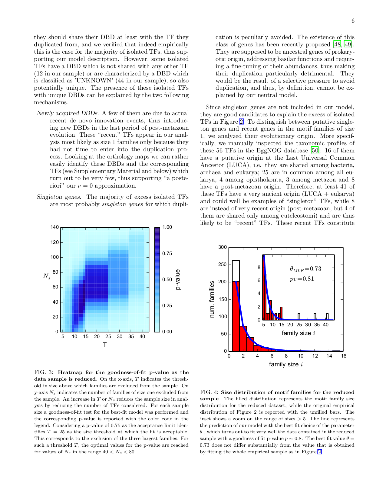 Transcription factors (TFs) exert their regulatory action by binding to DNA with specific sequence preferences. However, different TFs can partially share their binding sequences due to their common evolutionary origin. This `redundancy' of binding defines a way of organizing TFs in `motif families' by grouping TFs with similar binding preferences. Since these ultimately define the TF target genes, the motif family organization entails information about the structure of transcriptional regulation as it has been shaped by evolution. Focusing on the human TF repertoire, we show that a one-parameter evolutionary model of the Birth-Death-Innovation type can explain the TF empirical ripartition in motif families, and allows to highlight the relevant evolutionary forces at the origin of this organization. Moreover, the model allows to pinpoint few deviations from the neutral scenario it assumes: three over-expanded families (including HOX and FOX genes), a set of `singleton' TFs for which duplication seems to be selected against, and a higher-than-average rate of diversification of the binding preferences of TFs with a Zinc Finger DNA binding domain. Finally, a comparison of the TF motif family organization in different eukaryotic species suggests an increase of redundancy of binding with organism complexity.
Transcription factors (TFs) exert their regulatory action by binding to DNA with specific sequence preferences. However, different TFs can partially share their binding sequences due to their common evolutionary origin. This `redundancy' of binding defines a way of organizing TFs in `motif families' by grouping TFs with similar binding preferences. Since these ultimately define the TF target genes, the motif family organization entails information about the structure of transcriptional regulation as it has been shaped by evolution. Focusing on the human TF repertoire, we show that a one-parameter evolutionary model of the Birth-Death-Innovation type can explain the TF empirical ripartition in motif families, and allows to highlight the relevant evolutionary forces at the origin of this organization. Moreover, the model allows to pinpoint few deviations from the neutral scenario it assumes: three over-expanded families (including HOX and FOX genes), a set of `singleton' TFs for which duplication seems to be selected against, and a higher-than-average rate of diversification of the binding preferences of TFs with a Zinc Finger DNA binding domain. Finally, a comparison of the TF motif family organization in different eukaryotic species suggests an increase of redundancy of binding with organism complexity.

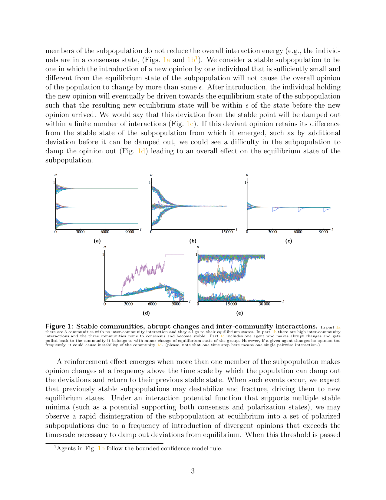
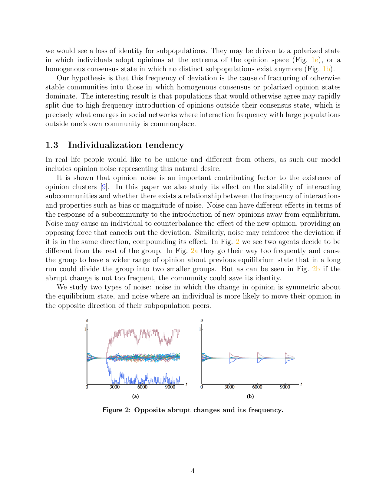

 Recent technological changes have increased connectivity between individuals around the world leading to higher frequency interactions between members of communities that would be otherwise distant and disconnected. This paper examines a model of opinion dynamics in interacting communities and studies how increasing interaction frequency affects the ability for communities to retain distinct identities versus falling into consensus or polarized states in which community identity is lost. We also study the effect (if any) of opinion noise related to a tendency for individuals to assert their individuality in homogenous populations. Our work builds on a model we developed previously [11] where the dynamics of opinion change is based on individual interactions that seek to minimize some energy potential based on the differences between opinions across the population.
Recent technological changes have increased connectivity between individuals around the world leading to higher frequency interactions between members of communities that would be otherwise distant and disconnected. This paper examines a model of opinion dynamics in interacting communities and studies how increasing interaction frequency affects the ability for communities to retain distinct identities versus falling into consensus or polarized states in which community identity is lost. We also study the effect (if any) of opinion noise related to a tendency for individuals to assert their individuality in homogenous populations. Our work builds on a model we developed previously [11] where the dynamics of opinion change is based on individual interactions that seek to minimize some energy potential based on the differences between opinions across the population.




 In this paper we propose a method to study a general vector-hosts mathematical model in order to explain how the changes in biodiversity could influence the dynamics of vector-borne diseases. We find that under the assumption of frequency-dependent transmission, i.e. the assumption that the number of contacts are diluted by the total population of hosts, the presence of a competent host is a necessary condition for the existence of an endemic state. In addition, we obtain that in the case of an endemic disease with a unique competent and resilient host, an increase in its density amplifies the disease.
In this paper we propose a method to study a general vector-hosts mathematical model in order to explain how the changes in biodiversity could influence the dynamics of vector-borne diseases. We find that under the assumption of frequency-dependent transmission, i.e. the assumption that the number of contacts are diluted by the total population of hosts, the presence of a competent host is a necessary condition for the existence of an endemic state. In addition, we obtain that in the case of an endemic disease with a unique competent and resilient host, an increase in its density amplifies the disease.




 We consider a class of birth-and-death processes describing a population made of $d$ sub-populations of different types which interact with one another. The state space is $\mathbb{Z}_+^d$ (unbounded). We assume that the population goes almost surely to extinction, so that the unique stationary distribution is the Dirac measure at the origin. These processes are parametrized by a scaling parameter $K$ which can be thought as the order of magnitude of the total size of the population at time $0$. For any fixed finite time span, it is well-known that such processes, when renormalized by $K$, are close, in the limit $K\to+\infty$, to the solutions of a certain differential equation in $\mathbb{R}_+^d$ whose vector field is determined by the birth and death rates. We consider the case where there is a unique attractive fixed point (off the boundary of the positive orthant) for the vector field (while the origin is repulsive). What is expected is that, for $K$ large, the process will stay in the vicinity of the fixed point for a very long time before being absorbed at the origin. To precisely describe this behavior, we prove the existence of a quasi-stationary distribution (qsd). In fact, we establish a bound for the total variation distance between the process conditioned to non-extinction before time $t$ and the qsd. This bound is exponentially small in $t$, for $t\gg \log K$. As a by-product, we obtain an estimate for the mean time to extinction in the qsd. We also quantify how close is the law of the process (not conditioned to non-extinction) either to the Dirac measure at the origin or to the qsd, for times much larger than $\log K$ and much smaller than the mean time to extinction, which is exponentially large as a function of $K$. Let us stress that we are interested in what happens for finite $K$. We obtain results much beyond what large deviation techniques could provide.
We consider a class of birth-and-death processes describing a population made of $d$ sub-populations of different types which interact with one another. The state space is $\mathbb{Z}_+^d$ (unbounded). We assume that the population goes almost surely to extinction, so that the unique stationary distribution is the Dirac measure at the origin. These processes are parametrized by a scaling parameter $K$ which can be thought as the order of magnitude of the total size of the population at time $0$. For any fixed finite time span, it is well-known that such processes, when renormalized by $K$, are close, in the limit $K\to+\infty$, to the solutions of a certain differential equation in $\mathbb{R}_+^d$ whose vector field is determined by the birth and death rates. We consider the case where there is a unique attractive fixed point (off the boundary of the positive orthant) for the vector field (while the origin is repulsive). What is expected is that, for $K$ large, the process will stay in the vicinity of the fixed point for a very long time before being absorbed at the origin. To precisely describe this behavior, we prove the existence of a quasi-stationary distribution (qsd). In fact, we establish a bound for the total variation distance between the process conditioned to non-extinction before time $t$ and the qsd. This bound is exponentially small in $t$, for $t\gg \log K$. As a by-product, we obtain an estimate for the mean time to extinction in the qsd. We also quantify how close is the law of the process (not conditioned to non-extinction) either to the Dirac measure at the origin or to the qsd, for times much larger than $\log K$ and much smaller than the mean time to extinction, which is exponentially large as a function of $K$. Let us stress that we are interested in what happens for finite $K$. We obtain results much beyond what large deviation techniques could provide.




 An elementary biostatistical theory based on a selectivity-variability principle is proposed to address a question raised by Charles Darwin, namely, how one sex of a sexually dimorphic species might tend to evolve with greater variability than the other sex. Briefly, the theory says that if one sex is relatively selective then from one generation to the next, more variable subpopulations of the opposite sex will generally tend to prevail over those with lesser variability. Moreover, the perhaps less intuitive converse also holds: if a sex is relatively non-selective, then less variable subpopulations of the opposite sex will prevail over those with greater variability. This theory requires certain regularity conditions on the distributions, but makes no assumptions about differences in means between the sexes, nor does it presume that one sex is selective and the other non-selective. Two mathematical models of the selectivity-variability principle are presented: a discrete-time one-step probabilistic model of short-term behavior with an example using normally distributed perceived fitness values; and a continuous-time deterministic model for the long-term asymptotic behavior of the expected sizes of the subpopulations with an example using exponentially distributed fitness levels.
An elementary biostatistical theory based on a selectivity-variability principle is proposed to address a question raised by Charles Darwin, namely, how one sex of a sexually dimorphic species might tend to evolve with greater variability than the other sex. Briefly, the theory says that if one sex is relatively selective then from one generation to the next, more variable subpopulations of the opposite sex will generally tend to prevail over those with lesser variability. Moreover, the perhaps less intuitive converse also holds: if a sex is relatively non-selective, then less variable subpopulations of the opposite sex will prevail over those with greater variability. This theory requires certain regularity conditions on the distributions, but makes no assumptions about differences in means between the sexes, nor does it presume that one sex is selective and the other non-selective. Two mathematical models of the selectivity-variability principle are presented: a discrete-time one-step probabilistic model of short-term behavior with an example using normally distributed perceived fitness values; and a continuous-time deterministic model for the long-term asymptotic behavior of the expected sizes of the subpopulations with an example using exponentially distributed fitness levels.



 The Moran model with recombination is considered, which describes the evolution of the genetic composition of a population under recombination and resampling. There are $n$ sites (or loci), a finite number of letters (or alleles) at every site, and we do not make any scaling assumptions. In particular, we do not assume a diffusion limit. We consider the following marginal ancestral recombination process. Let $S = \{1,...,n\}$ and $\mathcal A=\{A_1, ..., A_m\}$ be a partition of $S$. We concentrate on the joint probability of the letters at the sites in $A_1$ in individual $1$, $...$, and at the sites in $A_m$ in individual $m$, where the individuals are sampled from the current population without replacement. Following the ancestry of these sites backwards in time yields a process on the set of partitions of $S$, which, in the diffusion limit, turns into a marginalised version of the $n$-locus ancestral recombination graph. With the help of an inclusion-exclusion principle, we show that the type distribution corresponding to a given partition may be represented in a systematic way, with the help of so-called recombinators and sampling functions. The same is true of correlation functions (known as linkage disequilibria in genetics) of all orders. We prove that the partitioning process (backward in time) is dual to the Moran population process (forward in time), where the sampling function plays the role of the duality function. This sheds new light on the work of Bobrowski, Wojdyla, and Kimmel (2010). The result also leads to a closed system of ordinary differential equations for the expectations of the sampling functions, which can be translated into expected type distributions and expected linkage disequilibria.
The Moran model with recombination is considered, which describes the evolution of the genetic composition of a population under recombination and resampling. There are $n$ sites (or loci), a finite number of letters (or alleles) at every site, and we do not make any scaling assumptions. In particular, we do not assume a diffusion limit. We consider the following marginal ancestral recombination process. Let $S = \{1,...,n\}$ and $\mathcal A=\{A_1, ..., A_m\}$ be a partition of $S$. We concentrate on the joint probability of the letters at the sites in $A_1$ in individual $1$, $...$, and at the sites in $A_m$ in individual $m$, where the individuals are sampled from the current population without replacement. Following the ancestry of these sites backwards in time yields a process on the set of partitions of $S$, which, in the diffusion limit, turns into a marginalised version of the $n$-locus ancestral recombination graph. With the help of an inclusion-exclusion principle, we show that the type distribution corresponding to a given partition may be represented in a systematic way, with the help of so-called recombinators and sampling functions. The same is true of correlation functions (known as linkage disequilibria in genetics) of all orders. We prove that the partitioning process (backward in time) is dual to the Moran population process (forward in time), where the sampling function plays the role of the duality function. This sheds new light on the work of Bobrowski, Wojdyla, and Kimmel (2010). The result also leads to a closed system of ordinary differential equations for the expectations of the sampling functions, which can be translated into expected type distributions and expected linkage disequilibria.




 Topological phylogenetic trees can be assigned edge weights in several natural ways, highlighting different aspects of the tree. Here the rooted triple and quartet metrizations are introduced, and applied to formulate novel fast methods of inferring large trees from rooted triple and quartet data. These methods can be applied in new statistically consistent procedures for inference of a species tree from gene trees under the multispecies coalescent model.
Topological phylogenetic trees can be assigned edge weights in several natural ways, highlighting different aspects of the tree. Here the rooted triple and quartet metrizations are introduced, and applied to formulate novel fast methods of inferring large trees from rooted triple and quartet data. These methods can be applied in new statistically consistent procedures for inference of a species tree from gene trees under the multispecies coalescent model.

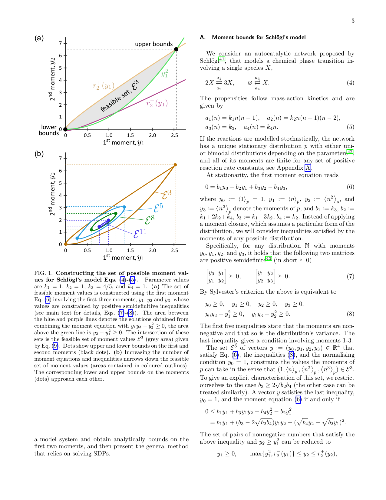
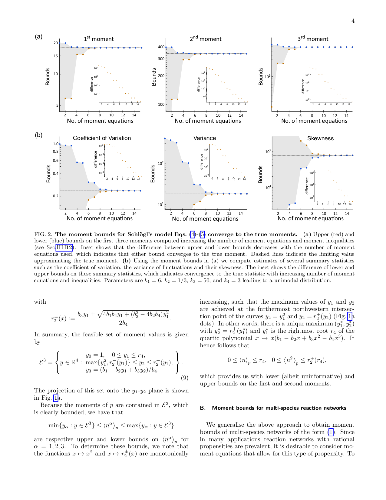

 The stochastic dynamics of biochemical networks are usually modelled with the chemical master equation (CME). The stationary distributions of CMEs are seldom solvable analytically, and numerical methods typically produce estimates with uncontrolled errors. Here, we introduce mathematical programming approaches that yield approximations of these distributions with computable error bounds which enable the verification of their accuracy. First, we use semidefinite programming to compute increasingly tighter upper and lower bounds on the moments of the stationary distributions for networks with rational propensities. Second, we use these moment bounds to formulate linear programs that yield convergent upper and lower bounds on the stationary distributions themselves, their marginals and stationary averages. The bounds obtained also provide a computational test for the uniqueness of the distribution. In the unique case, the bounds form an approximation of the stationary distribution with a computable bound on its error. In the non-unique case, our approach yields converging approximations of the ergodic distributions. We illustrate our methodology through several biochemical examples taken from the literature: Schl\"ogl's model for a chemical bifurcation, a two-dimensional toggle switch, a model for bursty gene expression, and a dimerisation model with multiple stationary distributions.
The stochastic dynamics of biochemical networks are usually modelled with the chemical master equation (CME). The stationary distributions of CMEs are seldom solvable analytically, and numerical methods typically produce estimates with uncontrolled errors. Here, we introduce mathematical programming approaches that yield approximations of these distributions with computable error bounds which enable the verification of their accuracy. First, we use semidefinite programming to compute increasingly tighter upper and lower bounds on the moments of the stationary distributions for networks with rational propensities. Second, we use these moment bounds to formulate linear programs that yield convergent upper and lower bounds on the stationary distributions themselves, their marginals and stationary averages. The bounds obtained also provide a computational test for the uniqueness of the distribution. In the unique case, the bounds form an approximation of the stationary distribution with a computable bound on its error. In the non-unique case, our approach yields converging approximations of the ergodic distributions. We illustrate our methodology through several biochemical examples taken from the literature: Schl\"ogl's model for a chemical bifurcation, a two-dimensional toggle switch, a model for bursty gene expression, and a dimerisation model with multiple stationary distributions.




 The spread of invasive species can have far reaching environmental and ecological consequences. Understanding invasion spread patterns and the underlying process driving invasions are key to predicting and managing invasions. We combine a set of statistical methods in a novel way to characterize local spread properties and demonstrate their application using simulated and historical data on invasive insects. Our method uses a Gaussian process fit to the surface of waiting times to invasion in order to characterize the vector field of spread. Using this method we estimate with statistical uncertainties the speed and direction of spread at each location. Simulations from a stratified diffusion model verify the accuracy of our method. We show how we may link local rates of spread to environmental covariates for two case studies: the spread of the gypsy moth (Lymantria dispar), and hemlock wolly adelgid (Adelges tsugae) in North America. We provide an R-package that automates the calculations for any spatially referenced waiting time data.
The spread of invasive species can have far reaching environmental and ecological consequences. Understanding invasion spread patterns and the underlying process driving invasions are key to predicting and managing invasions. We combine a set of statistical methods in a novel way to characterize local spread properties and demonstrate their application using simulated and historical data on invasive insects. Our method uses a Gaussian process fit to the surface of waiting times to invasion in order to characterize the vector field of spread. Using this method we estimate with statistical uncertainties the speed and direction of spread at each location. Simulations from a stratified diffusion model verify the accuracy of our method. We show how we may link local rates of spread to environmental covariates for two case studies: the spread of the gypsy moth (Lymantria dispar), and hemlock wolly adelgid (Adelges tsugae) in North America. We provide an R-package that automates the calculations for any spatially referenced waiting time data.




 One of the main aims of phylogenetics is to reconstruct the \enquote{Tree of Life}. In this respect, different methods and criteria are used to analyze DNA sequences of different species and to compare them in order to derive the evolutionary relationships of these species. Maximum Parsimony is one such criterion for tree reconstruction and, it is the one which we will use in this paper. However, it is well-known that tree reconstruction methods can lead to wrong relationship estimates. One typical problem of Maximum Parsimony is long branch attraction, which can lead to statistical inconsistency. In this work, we will consider a blockwise approach to alignment analysis, namely so-called $k$-tuple analyses. For four taxa it has already been shown that $k$-tuple-based analyses are statistically inconsistent if and only if the standard character-based (site-based) analyses are statistically inconsistent. So, in the four-taxon case, going from individual sites to $k$-tuples does not lead to any improvement. However, real biological analyses often consider more than only four taxa. Therefore, we analyze the case of five taxa for $2$- and $3$-tuple-site data and consider alphabets with two and four elements. We show that the equivalence of single-site data and $k$-tuple-site data then no longer holds. Even so, we can show that Maximum Parsimony is statistically inconsistent for $k$-tuple site data and five taxa.
One of the main aims of phylogenetics is to reconstruct the \enquote{Tree of Life}. In this respect, different methods and criteria are used to analyze DNA sequences of different species and to compare them in order to derive the evolutionary relationships of these species. Maximum Parsimony is one such criterion for tree reconstruction and, it is the one which we will use in this paper. However, it is well-known that tree reconstruction methods can lead to wrong relationship estimates. One typical problem of Maximum Parsimony is long branch attraction, which can lead to statistical inconsistency. In this work, we will consider a blockwise approach to alignment analysis, namely so-called $k$-tuple analyses. For four taxa it has already been shown that $k$-tuple-based analyses are statistically inconsistent if and only if the standard character-based (site-based) analyses are statistically inconsistent. So, in the four-taxon case, going from individual sites to $k$-tuples does not lead to any improvement. However, real biological analyses often consider more than only four taxa. Therefore, we analyze the case of five taxa for $2$- and $3$-tuple-site data and consider alphabets with two and four elements. We show that the equivalence of single-site data and $k$-tuple-site data then no longer holds. Even so, we can show that Maximum Parsimony is statistically inconsistent for $k$-tuple site data and five taxa.




 We investigate the global dynamics of a general Kermack-McKendrick-type epidemic model formulated in terms of a system of renewal equations. Specifically, we consider a renewal model for which both the force of infection and the infected removal rates are arbitrary functions of the infection age, $\tau$, and use the direct Lyapunov method to establish the global asymptotic stability of the equilibrium solutions. In particular, we show that the basic reproduction number, $R_0$, represents a sharp threshold parameter such that for $R_0\leq 1$, the infection-free equilibrium is globally asymptotically stable; whereas the endemic equilibrium becomes globally asymptotically stable when $R_0 > 1$, i.e. when it exists.
We investigate the global dynamics of a general Kermack-McKendrick-type epidemic model formulated in terms of a system of renewal equations. Specifically, we consider a renewal model for which both the force of infection and the infected removal rates are arbitrary functions of the infection age, $\tau$, and use the direct Lyapunov method to establish the global asymptotic stability of the equilibrium solutions. In particular, we show that the basic reproduction number, $R_0$, represents a sharp threshold parameter such that for $R_0\leq 1$, the infection-free equilibrium is globally asymptotically stable; whereas the endemic equilibrium becomes globally asymptotically stable when $R_0 > 1$, i.e. when it exists.