-
Background. A large number of algorithms is being developed to reconstruct
evolutionary models of individual tumours from genome sequencing data. Most
methods can analyze multiple samples collected either through bulk multi-region
sequencing experiments or the sequencing of individual cancer cells. However,
rarely the same method can support both data types.
Results. We introduce TRaIT, a computational framework to infer mutational
graphs that model the accumulation of multiple types of somatic alterations
driving tumour evolution. Compared to other tools, TRaIT supports multi-region
and single-cell sequencing data within the same statistical framework, and
delivers expressive models that capture many complex evolutionary phenomena.
TRaIT improves accuracy, robustness to data-specific errors and computational
complexity compared to competing methods.
Conclusions. We show that the application of TRaIT to single-cell and
multi-region cancer datasets can produce accurate and reliable models of
single-tumour evolution, quantify the extent of intra-tumour heterogeneity and
generate new testable experimental hypotheses.
-
Pathway Tools is a bioinformatics software environment with a broad set of
capabilities. The software provides genome-informatics tools such as a genome
browser, sequence alignments, a genome-variant analyzer, and
comparative-genomics operations. It offers metabolic-informatics tools, such as
metabolic reconstruction, quantitative metabolic modeling, prediction of
reaction atom mappings, and metabolic route search. Pathway Tools also provides
regulatory-informatics tools, such as the ability to represent and visualize a
wide range of regulatory interactions. The software creates and manages a type
of organism-specific database called a Pathway/Genome Database (PGDB), which
the software enables database curators to interactively edit. It supports web
publishing of PGDBs and provides a large number of query, visualization, and
omics-data analysis tools. Scientists around the world have created more than
9,800 PGDBs by using Pathway Tools, many of which are curated databases for
important model organisms. Those PGDBs can be exchanged using a peer-to-peer
database-sharing system called the PGDB Registry.
-
The advent of high--throughput transcription profiling technologies has
enabled identification of genes and pathways associated with disease, providing
new avenues for precision medicine. A key challenge is to analyze this data in
the context of the regulatory networks and pathways that control cellular
processes, while still obtaining insights that can be used to design new
diagnostic and therapeutic interventions. While classical differential
expression analysis provides specific and hence targetable gene-level insights,
it does not include any systems-level information. On the other hand, pathway
analyses integrate systems-level information with expression data, but are
often limited in their ability to indicate specific molecular targets. We
introduce GeneSurrounder, an analysis method that takes into account the
complex structure of interaction networks to identify specific genes that
disrupt pathway activity in a disease-specific manner. GeneSurrounder
integrates transcriptomic data and pathway network information in a novel
two-step procedure to detect genes that (i) appear to influence the expression
of other genes local to it in the network and (ii) are part of a subnetwork of
differentially expressed genes. Combined, this evidence can be used to pinpoint
specific genes that have a mechanistic role in the phenotype of interest.
Applying GeneSurrounder to three distinct ovarian cancer studies using a global
KEGG network, we show that our method is able to identify biologically relevant
genes and genes missed by single-gene association tests, integrate pathway and
expression data, and yield more consistent results across multiple studies of
the same phenotype than competing methods.
-
Transcription factors (TFs) exert their regulatory action by binding to DNA
with specific sequence preferences. However, different TFs can partially share
their binding sequences due to their common evolutionary origin. This
`redundancy' of binding defines a way of organizing TFs in `motif families' by
grouping TFs with similar binding preferences. Since these ultimately define
the TF target genes, the motif family organization entails information about
the structure of transcriptional regulation as it has been shaped by evolution.
Focusing on the human TF repertoire, we show that a one-parameter evolutionary
model of the Birth-Death-Innovation type can explain the TF empirical
ripartition in motif families, and allows to highlight the relevant
evolutionary forces at the origin of this organization. Moreover, the model
allows to pinpoint few deviations from the neutral scenario it assumes: three
over-expanded families (including HOX and FOX genes), a set of `singleton' TFs
for which duplication seems to be selected against, and a higher-than-average
rate of diversification of the binding preferences of TFs with a Zinc Finger
DNA binding domain. Finally, a comparison of the TF motif family organization
in different eukaryotic species suggests an increase of redundancy of binding
with organism complexity.
-
Motivated by the need for fast and accurate classification of unlabeled
nucleotide sequences on a large scale, we developed NASCUP, a new
classification method that captures statistical structures of nucleotide
sequences by compact context-tree models and universal probability from
information theory. NASCUP achieved BLAST-like classification accuracy
consistently for several large-scale databases in orders-of-magnitude reduced
runtime, and was applied to other bioinformatics tasks such as outlier
detection and synthetic sequence generation.
-
We introduce and study a set of training-free methods of
information-theoretic and algorithmic complexity nature applied to DNA
sequences to identify their potential capabilities to determine nucleosomal
binding sites. We test our measures on well-studied genomic sequences of
different sizes drawn from different sources. The measures reveal the known in
vivo versus in vitro predictive discrepancies and uncover their potential to
pinpoint (high) nucleosome occupancy. We explore different possible signals
within and beyond the nucleosome length and find that complexity indices are
informative of nucleosome occupancy. We compare against the gold standard
(Kaplan model) and find similar and complementary results with the main
difference that our sequence complexity approach. For example, for high
occupancy, complexity-based scores outperform the Kaplan model for predicting
binding representing a significant advancement in predicting the highest
nucleosome occupancy following a training-free approach.
-
We present ensemble methods in a machine learning (ML) framework combining
predictions from five known motif/binding site exploration algorithms. For a
given TF the ensemble starts with position weight matrices (PWM's) for the
motif, collected from the component algorithms. Using dimension reduction, we
identify significant PWM-based subspaces for analysis. Within each subspace a
machine classifier is built for identifying the TF's gene (promoter) targets
(Problem 1). These PWM-based subspaces form an ML-based sequence analysis tool.
Problem 2 (finding binding motifs) is solved by agglomerating k-mer (string)
feature PWM-based subspaces that stand out in identifying gene targets. We
approach Problem 3 (binding sites) with a novel machine learning approach that
uses promoter string features and ML importance scores in a classification
algorithm locating binding sites across the genome. For target gene
identification this method improves performance (measured by the F1 score) by
about 10 percentage points over the (a) motif scanning method and (b) the
coexpression-based association method. Top motif outperformed 5 component
algorithms as well as two other common algorithms (BEST and DEME). For
identifying individual binding sites on a benchmark cross species database
(Tompa et al., 2005) we match the best performer without much human
intervention. It also improved the performance on mammalian TFs.
The ensemble can integrate orthogonal information from different weak
learners (potentially using entirely different types of features) into a
machine learner that can perform consistently better for more TFs. The TF gene
target identification component (problem 1 above) is useful in constructing a
transcriptional regulatory network from known TF-target associations. The
ensemble is easily extendable to include more tools as well as future PWM-based
information.
-
The mainstream of research in genetics, epigenetics and imaging data analysis
focuses on statistical association or exploring statistical dependence between
variables. Despite their significant progresses in genetic research,
understanding the etiology and mechanism of complex phenotypes remains elusive.
Using association analysis as a major analytical platform for the complex data
analysis is a key issue that hampers the theoretic development of genomic
science and its application in practice. Causal inference is an essential
component for the discovery of mechanical relationships among complex
phenotypes. Many researchers suggest making the transition from association to
causation. Despite its fundamental role in science, engineering and
biomedicine, the traditional methods for causal inference require at least
three variables. However, quantitative genetic analysis such as QTL, eQTL,
mQTL, and genomic-imaging data analysis requires exploring the causal
relationships between two variables. This paper will focus on bivariate causal
discovery. We will introduce independence of cause and mechanism (ICM) as a
basic principle for causal inference, algorithmic information theory and
additive noise model (ANM) as major tools for bivariate causal discovery.
Large-scale simulations will be performed to evaluate the feasibility of the
ANM for bivariate causal discovery. To further evaluate their performance for
causal inference, the ANM will be applied to the construction of gene
regulatory networks. Also, the ANM will be applied to trait-imaging data
analysis to illustrate three scenarios: presence of both causation and
association, presence of association while absence of causation, and presence
of causation, while lack of association between two variables.
-
High quality gene models are necessary to expand the molecular and genetic
tools available for a target organism, but these are available for only a
handful of model organisms that have undergone extensive curation and
experimental validation over the course of many years. The majority of gene
models present in biological databases today have been identified in draft
genome assemblies using automated annotation pipelines that are frequently
based on orthologs from distantly related model organisms. Manual curation is
time consuming and often requires substantial expertise, but is instrumental in
improving gene model structure and identification. Manual annotation may seem
to be a daunting and cost-prohibitive task for small research communities but
involving undergraduates in community genome annotation consortiums can be
mutually beneficial for both education and improved genomic resources. We
outline a workflow for efficient manual annotation driven by a team of
primarily undergraduate annotators. This model can be scaled to large teams and
includes quality control processes through incremental evaluation. Moreover, it
gives students an opportunity to increase their understanding of genome biology
and to participate in scientific research in collaboration with peers and
senior researchers at multiple institutions.
-
Reaching the full potential of precision medicine depends on the quality of
personalized genome interpretation. In order to facilitate precision medicine
in regions of the Middle East and North Africa (MENA), a population-specific
reference genome for the indigenous Arab popula-tion of Qatar (QTRG) was
constructed by incorporating allele frequency data from sequencing of 1,161
Qataris, representing 0.4% of the population. A total of 20.9 million SNP and
3.1 million indels were observed in Qatar, including an average of 1.79% novel
variants per individual ge-nome. Replacement of the GRCh37 standard reference
with QTRG in a best practices genome analysis workflow resulted in an average
of 7* deeper coverage depth (an improvement of 23%), and 756,671 fewer variants
on average, a reduction of 16% that is attributed to common Qatari alleles
being present in the QTRG reference. The benefit for using QTRG varies across
ances-tries, a factor that should be taken into consideration when selecting an
appropriate reference for analysis.
-
Advancements in genomic research such as high-throughput sequencing
techniques have driven modern genomic studies into "big data" disciplines. This
data explosion is constantly challenging conventional methods used in genomics.
In parallel with the urgent demand for robust algorithms, deep learning has
succeeded in a variety of fields such as vision, speech, and text processing.
Yet genomics entails unique challenges to deep learning since we are expecting
from deep learning a superhuman intelligence that explores beyond our knowledge
to interpret the genome. A powerful deep learning model should rely on
insightful utilization of task-specific knowledge. In this paper, we briefly
discuss the strengths of different deep learning models from a genomic
perspective so as to fit each particular task with a proper deep architecture,
and remark on practical considerations of developing modern deep learning
architectures for genomics. We also provide a concise review of deep learning
applications in various aspects of genomic research, as well as pointing out
potential opportunities and obstacles for future genomics applications.
-
GeneVis is a web-based tool to visualize complementary data sets of different
disciplines within the field of genetics. It overlays gene-cluster information,
gene-interaction data and gene-disease association data by means of web-based
interactive graph visualizations. This allows an intuitive and quick assessment
of possible relations between the different datasets. By starting from a
high-level graph abstraction based on gene clusters, which can be selected for
detailed inspection at the gene-interaction level in a separate window, GeneVis
circumvents the common visual clutter problem when using gene datasets with a
high number of gene entries.
-
Sobottka and Hart (2011) made use of a Markovian concatenation model to
observe novel statistical symmetries in the mononucleotide and dinucleotide
distributions of a collection of bacterial chromosomes. The model roughly
approximates the first-order stochastic structure of DNA sequences by means of
a Markov chain whose one-step transition matrix is obtained from a
concatenation process guided by a positive persymmetric matrix $\mathfrak{L}$
of probabilities together with a positive parameter $m$. In this article we
carry out a detailed analysis of such stochastic matrices, here called
$\aleph$-generated matrices, and draw a number of conclusions. Necessary and
sufficient conditions for a stochastic matrix to be $\aleph$-generated are
given, as well as a way to determine if two $\aleph$-generated matrices can be
generated using the same persymmetric matrix with different values of $m$. The
results obtained in this research can be used to design new algorithms for
statistical analyses of real bacterial genomes.
-
Reconstruction of gene regulatory networks is the process of identifying gene
dependency from gene expression profile through some computation techniques. In
our human body, though all cells pose similar genetic material but the
activation state may vary. This variation in the activation of genes helps
researchers to understand more about the function of the cells. Researchers get
insight about diseases like mental illness, infectious disease, cancer disease
and heart disease from microarray technology, etc. In this study, a
cancer-specific gene regulatory network has been constructed using a simple and
novel machine learning approach. In First Step, linear regression algorithm
provided us the significant genes those expressed themselves differently. Next,
regulatory relationships between the identified genes has been computed using
Pearson correlation coefficient. Finally, the obtained results have been
validated with the available databases and literatures. We can identify the hub
genes and can be targeted for the cancer diagnosis.
-
Colletotrichum represent a genus of fungal species primarily known as plant
pathogens with severe economic impacts in temperate, subtropical and tropical
climates Consensus taxonomy and classification systems for Colletotrichum
species have been undergoing revision as high resolution genomic data becomes
available. Here we propose an alternative annotation that provides a complete
sequence for a Colletotrichum YPT1 gene homolog using the whole genome shotgun
sequence of Colletotrichum incanum isolated from soybean crops in Illinois,
USA.
-
Background: Since the invention of next-generation RNA sequencing (RNA-seq)
technologies, they have become a powerful tool to study the presence and
quantity of RNA molecules in biological samples and have revolutionized
transcriptomic studies. The analysis of RNA-seq data at four different levels
(samples, genes, transcripts, and exons) involve multiple statistical and
computational questions, some of which remain challenging up to date.
Results: We review RNA-seq analysis tools at the sample, gene, transcript,
and exon levels from a statistical perspective. We also highlight the
biological and statistical questions of most practical considerations.
Conclusion: The development of statistical and computational methods for
analyzing RNA- seq data has made significant advances in the past decade.
However, methods developed to answer the same biological question often rely on
diverse statical models and exhibit different performance under different
scenarios. This review discusses and compares multiple commonly used
statistical models regarding their assumptions, in the hope of helping users
select appropriate methods as needed, as well as assisting developers for
future method development.
-
Graphical models are powerful tools for modeling and making statistical
inferences regarding complex associations among variables in multivariate data.
In this paper we introduce the R package netgwas, which is designed based on
undirected graphical models to accomplish three important and interrelated
goals in genetics: constructing linkage map, reconstructing linkage
disequilibrium (LD) networks from multi-loci genotype data, and detecting
high-dimensional genotype-phenotype networks. The netgwas package deals with
species with any chromosome copy number in a unified way, unlike other
software. It implements recent improvements in both linkage map construction
(Behrouzi and Wit, 2018), and reconstructing conditional independence network
for non-Gaussian continuous data, discrete data, and mixed
discrete-and-continuous data (Behrouzi and Wit, 2017). Such datasets routinely
occur in genetics and genomics such as genotype data, and genotype-phenotype
data. We demonstrate the value of our package functionality by applying it to
various multivariate example datasets taken from the literature. We show, in
particular, that our package allows a more realistic analysis of data, as it
adjusts for the effect of all other variables while performing pairwise
associations. This feature controls for spurious associations between variables
that can arise from classical multiple testing approach. This paper includes a
brief overview of the statistical methods which have been implemented in the
package. The main body of the paper explains how to use the package. The
package uses a parallelization strategy on multi-core processors to speed-up
computations for large datasets. In addition, it contains several functions for
simulation and visualization. The netgwas package is freely available at
https://cran.r-project.org/web/packages/netgwas
-
Essential genes constitute the core of genes which cannot be mutated too much
nor lost along the evolutionary history of a species. Natural selection is
expected to be stricter on essential genes and on conserved (highly shared)
genes, than on genes that are either nonessential or peculiar to a single or a
few species. In order to further assess this expectation, we study here how
essentiality of a gene is connected with its degree of conservation among
several unrelated bacterial species, each one characterised by its own codon
usage bias. Confirming previous results on E. coli, we show the existence of a
universal exponential relation between gene essentiality and conservation in
bacteria. Moreover we show that, within each bacterial genome, there are at
least two groups of functionally distinct genes, characterised by different
levels of conservation and codon bias: i) a core of essential genes, mainly
related to cellular information processing; ii) a set of less conserved
nonessential genes with prevalent functions related to metabolism. In
particular, the genes in the first group are more retained among species, are
subject to a stronger purifying conservative selection and display a more
limited repertoire of synonymous codons. The core of essential genes is close
to the minimal bacterial genome, which is in the focus of recent studies in
synthetic biology, though we confirm that orthologs of genes that are essential
in one species are not necessarily essential in other species. We also list a
set of highly shared genes which, reasonably, could constitute a reservoir of
targets for new anti-microbial drugs.
-
A large number of recent genome-wide association studies (GWASs) for complex
phenotypes confirm the early conjecture for polygenicity, suggesting the
presence of large number of variants with only tiny or moderate effects.
However, due to the limited sample size of a single GWAS, many associated
genetic variants are too weak to achieve the genome-wide significance. These
undiscovered variants further limit the prediction capability of GWAS.
Restricted access to the individual-level data and the increasing availability
of the published GWAS results motivate the development of methods integrating
both the individual-level and summary-level data. How to build the connection
between the individual-level and summary-level data determines the efficiency
of using the existing abundant summary-level resources with limited
individual-level data, and this issue inspires more efforts in the existing
area.
In this study, we propose a novel statistical approach, LEP, which provides a
novel way of modeling the connection between the individual-level data and
summary-level data. LEP integrates both types of data by \underline{LE}veraing
\underline{P}leiotropy to increase the statistical power of risk variants
identification and the accuracy of risk prediction. The algorithm for parameter
estimation is developed to handle genome-wide-scale data. Through comprehensive
simulation studies, we demonstrated the advantages of LEP over the existing
methods. We further applied LEP to perform integrative analysis of Crohn's
disease from WTCCC and summary statistics from GWAS of some other diseases,
such as Type 1 diabetes, Ulcerative colitis and Primary biliary cirrhosis. LEP
was able to significantly increase the statistical power of identifying risk
variants and improve the risk prediction accuracy from 63.39\% ($\pm$ 0.58\%)
to 68.33\% ($\pm$ 0.32\%) using about 195,000 variants.
-
Aligning millions of short DNA or RNA reads, of 75 to 250 base pairs each, to
a reference genome is a significant computation problem in bioinformatics. We
present a flexible and fast FPGA-based short read alignment tool. Our aligner
makes use of the processing power of FPGAs in conjunction with the greater host
memory bandwidth and flexibility of software to improve performance and achieve
a high level of configurability. This flexible design supports a variety of
reference genome sizes without the performance degradation suffered by other
software and FPGA-based aligners. It is also better able to support the
features of new alignment algorithms, which frequently crop up in the rapidly
evolving field of bioinformatics. We demonstrate these advantages in a case
study where we align RNA-Seq data from a hypothesized mouse / human xenograft.
In this case study, our aligner provides a speedup of 5.6x over BWA-SW with
energy savings of 21%, while also reducing incorrect short read classification
by 29%. To demonstrate the flexibility of our system we show that the speedup
can be substantially improved while retaining most of the accuracy gains over
BWA-SW. The speedup can be increased to 71.3x, while still enjoying a 28%
incorrect classification improvement and 52% improvement in unaligned reads.
-
Many complex systems are modular. Such systems can be represented as
"component systems", i.e., sets of elementary components, such as LEGO bricks
in LEGO sets. The bricks found in a LEGO set reflect a target architecture,
which can be built following a set-specific list of instructions. In other
component systems, instead, the underlying functional design and constraints
are not obvious a priori, and their detection is often a challenge of both
scientific and practical importance, requiring a clear understanding of
component statistics. Importantly, some quantitative invariants appear to be
common to many component systems, most notably a common broad distribution of
component abundances, which often resembles the well-known Zipf's law. Such
"laws" affect in a general and non-trivial way the component statistics,
potentially hindering the identification of system-specific functional
constraints or generative processes. Here, we specifically focus on the
statistics of shared components, i.e., the distribution of the number of
components shared by different system-realizations, such as the common bricks
found in different LEGO sets. To account for the effects of component
heterogeneity, we consider a simple null model, which builds
system-realizations by random draws from a universe of possible components.
Under general assumptions on abundance heterogeneity, we provide analytical
estimates of component occurrence, which quantify exhaustively the statistics
of shared components. Surprisingly, this simple null model can positively
explain important features of empirical component-occurrence distributions
obtained from data on bacterial genomes, LEGO sets, and book chapters. Specific
architectural features and functional constraints can be detected from
occurrence patterns as deviations from these null predictions, as we show for
the illustrative case of the "core" genome in bacteria.
-
Recently, it has become feasible to generate large-scale, multi-tissue gene
expression data, where expression profiles are obtained from multiple tissues
or organs sampled from dozens to hundreds of individuals. When traditional
clustering methods are applied to this type of data, important information is
lost, because they either require all tissues to be analyzed independently,
ignoring dependencies and similarities between tissues, or to merge tissues in
a single, monolithic dataset, ignoring individual characteristics of tissues.
We developed a Bayesian model-based multi-tissue clustering algorithm, revamp,
which can incorporate prior information on physiological tissue similarity, and
which results in a set of clusters, each consisting of a core set of genes
conserved across tissues as well as differential sets of genes specific to one
or more subsets of tissues. Using data from seven vascular and metabolic
tissues from over 100 individuals in the STockholm Atherosclerosis Gene
Expression (STAGE) study, we demonstrate that multi-tissue clusters inferred by
revamp are more enriched for tissue-dependent protein-protein interactions
compared to alternative approaches. We further demonstrate that revamp results
in easily interpretable multi-tissue gene expression associations to key
coronary artery disease processes and clinical phenotypes in the STAGE
individuals. Revamp is implemented in the Lemon-Tree software, available at
https://github.com/eb00/lemon-tree
-
The aetiology of polygenic obesity is multifactorial, which indicates that
life-style and environmental factors may influence multiples genes to aggravate
this disorder. Several low-risk single nucleotide polymorphisms (SNPs) have
been associated with BMI. However, identified loci only explain a small
proportion of the variation ob-served for this phenotype. The linear nature of
genome wide association studies (GWAS) used to identify associations between
genetic variants and the phenotype have had limited success in explaining the
heritability variation of BMI and shown low predictive capacity in
classification studies. GWAS ignores the epistatic interactions that less
significant variants have on the phenotypic outcome. In this paper we utilise a
novel deep learning-based methodology to reduce the high dimensional space in
GWAS and find epistatic interactions between SNPs for classification purposes.
SNPs were filtered based on the effects associations have with BMI. Since
Bonferroni adjustment for multiple testing is highly conservative, an important
proportion of SNPs involved in SNP-SNP interactions are ignored. Therefore,
only SNPs with p-values < 1x10-2 were considered for subsequent epistasis
analysis using stacked auto encoders (SAE). This allows the nonlinearity
present in SNP-SNP interactions to be discovered through progressively smaller
hidden layer units and to initialise a multi-layer feedforward artificial
neural network (ANN) classifier. The classifier is fine-tuned to classify
extremely obese and non-obese individuals. The best results were obtained with
2000 compressed units (SE=0.949153, SP=0.933014, Gini=0.949936,
Lo-gloss=0.1956, AUC=0.97497 and MSE=0.054057). Using 50 compressed units it
was possible to achieve (SE=0.785311, SP=0.799043, Gini=0.703566,
Logloss=0.476864, AUC=0.85178 and MSE=0.156315).
-
DNA methylation is a well-studied genetic modification crucial to regulate
the functioning of the genome. Its alterations play an important role in
tumorigenesis and tumor-suppression. Thus, studying DNA methylation data may
help biomarker discovery in cancer. Since public data on DNA methylation become
abundant, and considering the high number of methylated sites (features)
present in the genome, it is important to have a method for efficiently
processing such large datasets. Relying on big data technologies, we propose
BIGBIOCL an algorithm that can apply supervised classification methods to
datasets with hundreds of thousands of features. It is designed for the
extraction of alternative and equivalent classification models through
iterative deletion of selected features. We run experiments on DNA methylation
datasets extracted from The Cancer Genome Atlas, focusing on three tumor types:
breast, kidney, and thyroid carcinomas. We perform classifications extracting
several methylated sites and their associated genes with accurate performance.
Results suggest that BIGBIOCL can perform hundreds of classification iterations
on hundreds of thousands of features in few hours. Moreover, we compare the
performance of our method with other state-of-the-art classifiers and with a
wide-spread DNA methylation analysis method based on network analysis. Finally,
we are able to efficiently compute multiple alternative classification models
and extract, from DNA-methylation large datasets, a set of candidate genes to
be further investigated to determine their active role in cancer. BIGBIOCL,
results of experiments, and a guide to carry on new experiments are freely
available on GitHub.
-
In this paper, association results from genome-wide association studies
(GWAS) are combined with a deep learning framework to test the predictive
capacity of statistically significant single nucleotide polymorphism (SNPs)
associated with obesity phenotype. Our approach demonstrates the potential of
deep learning as a powerful framework for GWAS analysis that can capture
information about SNPs and the important interactions between them. Basic
statistical methods and techniques for the analysis of genetic SNP data from
population-based genome-wide studies have been considered. Statistical
association testing between individual SNPs and obesity was conducted under an
additive model using logistic regression. Four subsets of loci after
quality-control (QC) and association analysis were selected: P-values lower
than 1x10-5 (5 SNPs), 1x10-4 (32 SNPs), 1x10-3 (248 SNPs) and 1x10-2 (2465
SNPs). A deep learning classifier is initialised using these sets of SNPs and
fine-tuned to classify obese and non-obese observations. Using a deep learning
classifier model and genetic variants with P-value < 1x10-2 (2465 SNPs) it was
possible to obtain results (SE=0.9604, SP=0.9712, Gini=0.9817, LogLoss=0.1150,
AUC=0.9908 and MSE=0.0300). As the P-value increased, an evident deterioration
in performance was observed. Results demonstrate that single SNP analysis fails
to capture the cumulative effect of less significant variants and their overall
contribution to the outcome in disease prediction, which is captured using a
deep learning framework.





 Pathway Tools is a bioinformatics software environment with a broad set of capabilities. The software provides genome-informatics tools such as a genome browser, sequence alignments, a genome-variant analyzer, and comparative-genomics operations. It offers metabolic-informatics tools, such as metabolic reconstruction, quantitative metabolic modeling, prediction of reaction atom mappings, and metabolic route search. Pathway Tools also provides regulatory-informatics tools, such as the ability to represent and visualize a wide range of regulatory interactions. The software creates and manages a type of organism-specific database called a Pathway/Genome Database (PGDB), which the software enables database curators to interactively edit. It supports web publishing of PGDBs and provides a large number of query, visualization, and omics-data analysis tools. Scientists around the world have created more than 9,800 PGDBs by using Pathway Tools, many of which are curated databases for important model organisms. Those PGDBs can be exchanged using a peer-to-peer database-sharing system called the PGDB Registry.
Pathway Tools is a bioinformatics software environment with a broad set of capabilities. The software provides genome-informatics tools such as a genome browser, sequence alignments, a genome-variant analyzer, and comparative-genomics operations. It offers metabolic-informatics tools, such as metabolic reconstruction, quantitative metabolic modeling, prediction of reaction atom mappings, and metabolic route search. Pathway Tools also provides regulatory-informatics tools, such as the ability to represent and visualize a wide range of regulatory interactions. The software creates and manages a type of organism-specific database called a Pathway/Genome Database (PGDB), which the software enables database curators to interactively edit. It supports web publishing of PGDBs and provides a large number of query, visualization, and omics-data analysis tools. Scientists around the world have created more than 9,800 PGDBs by using Pathway Tools, many of which are curated databases for important model organisms. Those PGDBs can be exchanged using a peer-to-peer database-sharing system called the PGDB Registry.




 The advent of high--throughput transcription profiling technologies has enabled identification of genes and pathways associated with disease, providing new avenues for precision medicine. A key challenge is to analyze this data in the context of the regulatory networks and pathways that control cellular processes, while still obtaining insights that can be used to design new diagnostic and therapeutic interventions. While classical differential expression analysis provides specific and hence targetable gene-level insights, it does not include any systems-level information. On the other hand, pathway analyses integrate systems-level information with expression data, but are often limited in their ability to indicate specific molecular targets. We introduce GeneSurrounder, an analysis method that takes into account the complex structure of interaction networks to identify specific genes that disrupt pathway activity in a disease-specific manner. GeneSurrounder integrates transcriptomic data and pathway network information in a novel two-step procedure to detect genes that (i) appear to influence the expression of other genes local to it in the network and (ii) are part of a subnetwork of differentially expressed genes. Combined, this evidence can be used to pinpoint specific genes that have a mechanistic role in the phenotype of interest. Applying GeneSurrounder to three distinct ovarian cancer studies using a global KEGG network, we show that our method is able to identify biologically relevant genes and genes missed by single-gene association tests, integrate pathway and expression data, and yield more consistent results across multiple studies of the same phenotype than competing methods.
The advent of high--throughput transcription profiling technologies has enabled identification of genes and pathways associated with disease, providing new avenues for precision medicine. A key challenge is to analyze this data in the context of the regulatory networks and pathways that control cellular processes, while still obtaining insights that can be used to design new diagnostic and therapeutic interventions. While classical differential expression analysis provides specific and hence targetable gene-level insights, it does not include any systems-level information. On the other hand, pathway analyses integrate systems-level information with expression data, but are often limited in their ability to indicate specific molecular targets. We introduce GeneSurrounder, an analysis method that takes into account the complex structure of interaction networks to identify specific genes that disrupt pathway activity in a disease-specific manner. GeneSurrounder integrates transcriptomic data and pathway network information in a novel two-step procedure to detect genes that (i) appear to influence the expression of other genes local to it in the network and (ii) are part of a subnetwork of differentially expressed genes. Combined, this evidence can be used to pinpoint specific genes that have a mechanistic role in the phenotype of interest. Applying GeneSurrounder to three distinct ovarian cancer studies using a global KEGG network, we show that our method is able to identify biologically relevant genes and genes missed by single-gene association tests, integrate pathway and expression data, and yield more consistent results across multiple studies of the same phenotype than competing methods.


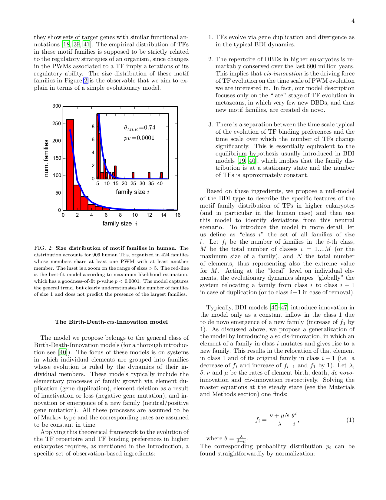

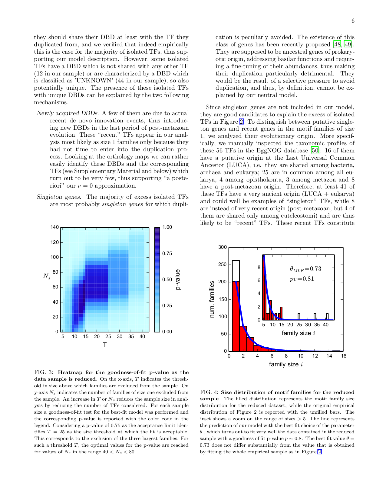 Transcription factors (TFs) exert their regulatory action by binding to DNA with specific sequence preferences. However, different TFs can partially share their binding sequences due to their common evolutionary origin. This `redundancy' of binding defines a way of organizing TFs in `motif families' by grouping TFs with similar binding preferences. Since these ultimately define the TF target genes, the motif family organization entails information about the structure of transcriptional regulation as it has been shaped by evolution. Focusing on the human TF repertoire, we show that a one-parameter evolutionary model of the Birth-Death-Innovation type can explain the TF empirical ripartition in motif families, and allows to highlight the relevant evolutionary forces at the origin of this organization. Moreover, the model allows to pinpoint few deviations from the neutral scenario it assumes: three over-expanded families (including HOX and FOX genes), a set of `singleton' TFs for which duplication seems to be selected against, and a higher-than-average rate of diversification of the binding preferences of TFs with a Zinc Finger DNA binding domain. Finally, a comparison of the TF motif family organization in different eukaryotic species suggests an increase of redundancy of binding with organism complexity.
Transcription factors (TFs) exert their regulatory action by binding to DNA with specific sequence preferences. However, different TFs can partially share their binding sequences due to their common evolutionary origin. This `redundancy' of binding defines a way of organizing TFs in `motif families' by grouping TFs with similar binding preferences. Since these ultimately define the TF target genes, the motif family organization entails information about the structure of transcriptional regulation as it has been shaped by evolution. Focusing on the human TF repertoire, we show that a one-parameter evolutionary model of the Birth-Death-Innovation type can explain the TF empirical ripartition in motif families, and allows to highlight the relevant evolutionary forces at the origin of this organization. Moreover, the model allows to pinpoint few deviations from the neutral scenario it assumes: three over-expanded families (including HOX and FOX genes), a set of `singleton' TFs for which duplication seems to be selected against, and a higher-than-average rate of diversification of the binding preferences of TFs with a Zinc Finger DNA binding domain. Finally, a comparison of the TF motif family organization in different eukaryotic species suggests an increase of redundancy of binding with organism complexity.




 Motivated by the need for fast and accurate classification of unlabeled nucleotide sequences on a large scale, we developed NASCUP, a new classification method that captures statistical structures of nucleotide sequences by compact context-tree models and universal probability from information theory. NASCUP achieved BLAST-like classification accuracy consistently for several large-scale databases in orders-of-magnitude reduced runtime, and was applied to other bioinformatics tasks such as outlier detection and synthetic sequence generation.
Motivated by the need for fast and accurate classification of unlabeled nucleotide sequences on a large scale, we developed NASCUP, a new classification method that captures statistical structures of nucleotide sequences by compact context-tree models and universal probability from information theory. NASCUP achieved BLAST-like classification accuracy consistently for several large-scale databases in orders-of-magnitude reduced runtime, and was applied to other bioinformatics tasks such as outlier detection and synthetic sequence generation.



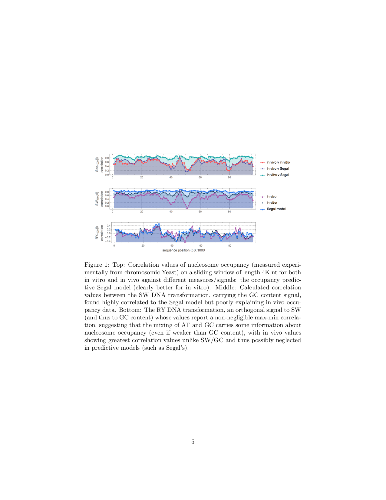
 We introduce and study a set of training-free methods of information-theoretic and algorithmic complexity nature applied to DNA sequences to identify their potential capabilities to determine nucleosomal binding sites. We test our measures on well-studied genomic sequences of different sizes drawn from different sources. The measures reveal the known in vivo versus in vitro predictive discrepancies and uncover their potential to pinpoint (high) nucleosome occupancy. We explore different possible signals within and beyond the nucleosome length and find that complexity indices are informative of nucleosome occupancy. We compare against the gold standard (Kaplan model) and find similar and complementary results with the main difference that our sequence complexity approach. For example, for high occupancy, complexity-based scores outperform the Kaplan model for predicting binding representing a significant advancement in predicting the highest nucleosome occupancy following a training-free approach.
We introduce and study a set of training-free methods of information-theoretic and algorithmic complexity nature applied to DNA sequences to identify their potential capabilities to determine nucleosomal binding sites. We test our measures on well-studied genomic sequences of different sizes drawn from different sources. The measures reveal the known in vivo versus in vitro predictive discrepancies and uncover their potential to pinpoint (high) nucleosome occupancy. We explore different possible signals within and beyond the nucleosome length and find that complexity indices are informative of nucleosome occupancy. We compare against the gold standard (Kaplan model) and find similar and complementary results with the main difference that our sequence complexity approach. For example, for high occupancy, complexity-based scores outperform the Kaplan model for predicting binding representing a significant advancement in predicting the highest nucleosome occupancy following a training-free approach.

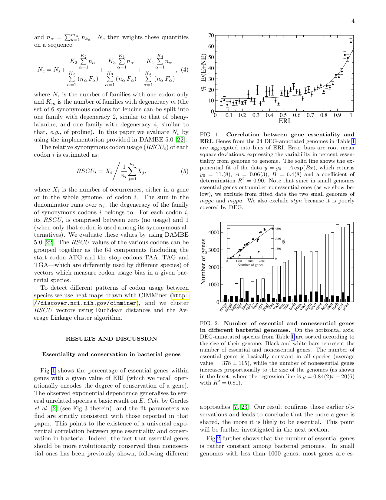

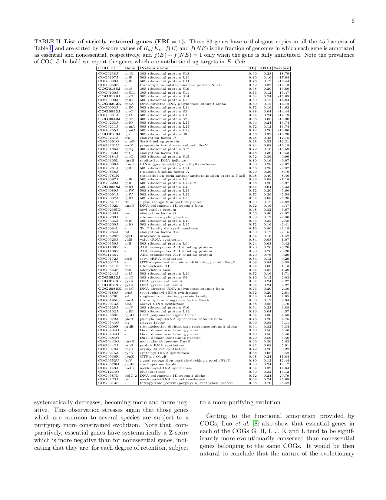 Essential genes constitute the core of genes which cannot be mutated too much nor lost along the evolutionary history of a species. Natural selection is expected to be stricter on essential genes and on conserved (highly shared) genes, than on genes that are either nonessential or peculiar to a single or a few species. In order to further assess this expectation, we study here how essentiality of a gene is connected with its degree of conservation among several unrelated bacterial species, each one characterised by its own codon usage bias. Confirming previous results on E. coli, we show the existence of a universal exponential relation between gene essentiality and conservation in bacteria. Moreover we show that, within each bacterial genome, there are at least two groups of functionally distinct genes, characterised by different levels of conservation and codon bias: i) a core of essential genes, mainly related to cellular information processing; ii) a set of less conserved nonessential genes with prevalent functions related to metabolism. In particular, the genes in the first group are more retained among species, are subject to a stronger purifying conservative selection and display a more limited repertoire of synonymous codons. The core of essential genes is close to the minimal bacterial genome, which is in the focus of recent studies in synthetic biology, though we confirm that orthologs of genes that are essential in one species are not necessarily essential in other species. We also list a set of highly shared genes which, reasonably, could constitute a reservoir of targets for new anti-microbial drugs.
Essential genes constitute the core of genes which cannot be mutated too much nor lost along the evolutionary history of a species. Natural selection is expected to be stricter on essential genes and on conserved (highly shared) genes, than on genes that are either nonessential or peculiar to a single or a few species. In order to further assess this expectation, we study here how essentiality of a gene is connected with its degree of conservation among several unrelated bacterial species, each one characterised by its own codon usage bias. Confirming previous results on E. coli, we show the existence of a universal exponential relation between gene essentiality and conservation in bacteria. Moreover we show that, within each bacterial genome, there are at least two groups of functionally distinct genes, characterised by different levels of conservation and codon bias: i) a core of essential genes, mainly related to cellular information processing; ii) a set of less conserved nonessential genes with prevalent functions related to metabolism. In particular, the genes in the first group are more retained among species, are subject to a stronger purifying conservative selection and display a more limited repertoire of synonymous codons. The core of essential genes is close to the minimal bacterial genome, which is in the focus of recent studies in synthetic biology, though we confirm that orthologs of genes that are essential in one species are not necessarily essential in other species. We also list a set of highly shared genes which, reasonably, could constitute a reservoir of targets for new anti-microbial drugs.

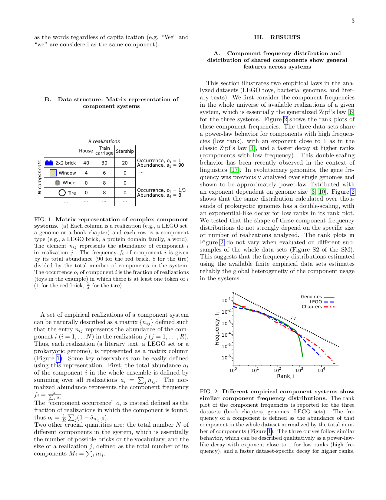
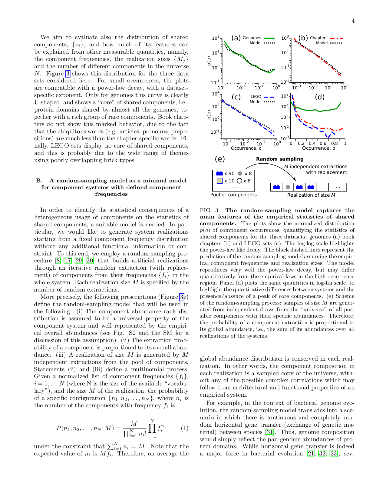

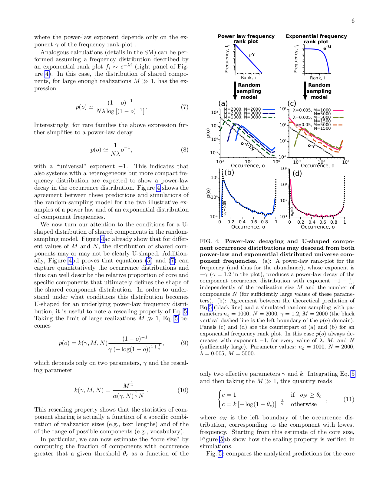 Many complex systems are modular. Such systems can be represented as "component systems", i.e., sets of elementary components, such as LEGO bricks in LEGO sets. The bricks found in a LEGO set reflect a target architecture, which can be built following a set-specific list of instructions. In other component systems, instead, the underlying functional design and constraints are not obvious a priori, and their detection is often a challenge of both scientific and practical importance, requiring a clear understanding of component statistics. Importantly, some quantitative invariants appear to be common to many component systems, most notably a common broad distribution of component abundances, which often resembles the well-known Zipf's law. Such "laws" affect in a general and non-trivial way the component statistics, potentially hindering the identification of system-specific functional constraints or generative processes. Here, we specifically focus on the statistics of shared components, i.e., the distribution of the number of components shared by different system-realizations, such as the common bricks found in different LEGO sets. To account for the effects of component heterogeneity, we consider a simple null model, which builds system-realizations by random draws from a universe of possible components. Under general assumptions on abundance heterogeneity, we provide analytical estimates of component occurrence, which quantify exhaustively the statistics of shared components. Surprisingly, this simple null model can positively explain important features of empirical component-occurrence distributions obtained from data on bacterial genomes, LEGO sets, and book chapters. Specific architectural features and functional constraints can be detected from occurrence patterns as deviations from these null predictions, as we show for the illustrative case of the "core" genome in bacteria.
Many complex systems are modular. Such systems can be represented as "component systems", i.e., sets of elementary components, such as LEGO bricks in LEGO sets. The bricks found in a LEGO set reflect a target architecture, which can be built following a set-specific list of instructions. In other component systems, instead, the underlying functional design and constraints are not obvious a priori, and their detection is often a challenge of both scientific and practical importance, requiring a clear understanding of component statistics. Importantly, some quantitative invariants appear to be common to many component systems, most notably a common broad distribution of component abundances, which often resembles the well-known Zipf's law. Such "laws" affect in a general and non-trivial way the component statistics, potentially hindering the identification of system-specific functional constraints or generative processes. Here, we specifically focus on the statistics of shared components, i.e., the distribution of the number of components shared by different system-realizations, such as the common bricks found in different LEGO sets. To account for the effects of component heterogeneity, we consider a simple null model, which builds system-realizations by random draws from a universe of possible components. Under general assumptions on abundance heterogeneity, we provide analytical estimates of component occurrence, which quantify exhaustively the statistics of shared components. Surprisingly, this simple null model can positively explain important features of empirical component-occurrence distributions obtained from data on bacterial genomes, LEGO sets, and book chapters. Specific architectural features and functional constraints can be detected from occurrence patterns as deviations from these null predictions, as we show for the illustrative case of the "core" genome in bacteria.