-
Due to the scarcity of quantitative details about biological phenomena,
quantitative modeling in systems biology can be compromised, especially at the
subcellular scale. One way to get around this is qualitative modeling because
it requires few to no quantitative information. One of the most popular
qualitative modeling approaches is the Boolean network formalism. However,
Boolean models allow variables to take only two values, which can be too
simplistic in some cases. The present work proposes a modeling approach derived
from Boolean networks where continuous logical operators are used and where
edges can be tuned. Using continuous logical operators allows variables to be
more finely valued while remaining qualitative. To consider that some
biological interactions can be slower or weaker than other ones, edge states
are also computed in order to modulate in speed and strength the signal they
convey. The proposed formalism is illustrated on a toy network coming from the
epidermal growth factor receptor signaling pathway. The obtained simulations
show that continuous results are produced, thus allowing finer analysis. The
simulations also show that modulating the signal conveyed by the edges allows
to incorporate knowledge about the interactions they model. The goal is to
provide enhancements in the ability of qualitative models to simulate the
dynamics of biological networks while limiting the need of quantitative
information.
-
Estimation of the number of species or unobserved classes from a random
sample of the underlying population is a ubiquitous problem in statistics. In
classical settings, the size of the sample is usually small. New technologies
such as high-throughput DNA sequencing have allowed for the sampling of
extremely large and heterogeneous populations at scales not previously
attainable or even considered. New algorithms are required that take advantage
of the size of the data to account for heterogeneity, but are also sufficiently
fast and scale well with large data. We present a non-parametric moment-based
estimator that is both computationally efficient and is sufficiently flexible
to account for heterogeneity in the abundances of underlying population. This
estimator is based on an extension of a popular moment-based lower bound (Chao,
1984), originally developed by Harris (1959) but unattainable due to the lack
of economical algorithms to solve the system of nonlinear equation required for
estimation. We apply results from the classical moment problem to show that
solutions can be obtained efficiently, allowing for estimators that are
simultaneously conservative and use more information. This is critical for
modern genomic applications, where there may be many large experiments that
require the application of species estimation. We present applications of our
estimator to estimating T-Cell receptor repertoire and dropout in single cell
RNA-seq experiments.
-
In usual demographic analysis, force of mortality is a function of one
variable, that is, of age. In this article bi-variate and multivariate force of
mortality functions are introduced for the first time to explain mortality
differentials. The pattern of mortality in a population is one of the strong
influencing factors in determining the life expectancies at various ages in the
population. Considering univariate functions of age only to understand the
human mortality data without associating with other variables could lead to
incomplete analysis. The reasons behind declining forces of mortality globally
could be studied using the proposed functions. Other applications of
multivariate forces of mortality could be in actuarial sciences.
-
Cryo-electron microscopy provides 2-D projection images of the 3-D electron
scattering intensity of many instances of the particle under study (e.g., a
virus). Both symmetry (rotational point groups) and heterogeneity are important
aspects of biological particles and both aspects can be combined by describing
the electron scattering intensity of the particle as a stochastic process with
a symmetric probability law and therefore symmetric moments. A maximum
likelihood estimator implemented by an expectation-maximization algorithm is
described which estimates the unknown statistics of the electron scattering
intensity stochastic process from images of instances of the particle. The
algorithm is demonstrated on the bacteriophage HK97 and the virus N$\omega$V.
The results are contrasted with existing algorithms which assume that each
instance of the particle has the symmetry rather than the less restrictive
assumption that the probability law has the symmetry.
-
We introduce a tensor-based clustering method to extract sparse,
low-dimensional structure from high-dimensional, multi-indexed datasets. This
framework is designed to enable detection of clusters of data in the presence
of structural requirements which we encode as algebraic constraints in a linear
program. Our clustering method is general and can be tailored to a variety of
applications in science and industry. We illustrate our method on a collection
of experiments measuring the response of genetically diverse breast cancer cell
lines to an array of ligands. Each experiment consists of a cell line-ligand
combination, and contains time-course measurements of the early-signalling
kinases MAPK and AKT at two different ligand dose levels. By imposing
appropriate structural constraints and respecting the multi-indexed structure
of the data, the analysis of clusters can be optimized for biological
interpretation and therapeutic understanding. We then perform a systematic,
large-scale exploration of mechanistic models of MAPK-AKT crosstalk for each
cluster. This analysis allows us to quantify the heterogeneity of breast cancer
cell subtypes, and leads to hypotheses about the signalling mechanisms that
mediate the response of the cell lines to ligands.
-
Pathway Tools is a bioinformatics software environment with a broad set of
capabilities. The software provides genome-informatics tools such as a genome
browser, sequence alignments, a genome-variant analyzer, and
comparative-genomics operations. It offers metabolic-informatics tools, such as
metabolic reconstruction, quantitative metabolic modeling, prediction of
reaction atom mappings, and metabolic route search. Pathway Tools also provides
regulatory-informatics tools, such as the ability to represent and visualize a
wide range of regulatory interactions. The software creates and manages a type
of organism-specific database called a Pathway/Genome Database (PGDB), which
the software enables database curators to interactively edit. It supports web
publishing of PGDBs and provides a large number of query, visualization, and
omics-data analysis tools. Scientists around the world have created more than
9,800 PGDBs by using Pathway Tools, many of which are curated databases for
important model organisms. Those PGDBs can be exchanged using a peer-to-peer
database-sharing system called the PGDB Registry.
-
The development of chemical reaction models aids understanding and prediction
in areas ranging from biology to electrochemistry and combustion. A systematic
approach to building reaction network models uses observational data not only
to estimate unknown parameters, but also to learn model structure. Bayesian
inference provides a natural approach to this data-driven construction of
models. Yet traditional Bayesian model inference methodologies that numerically
evaluate the evidence for each model are often infeasible for nonlinear
reaction network inference, as the number of plausible models can be
combinatorially large. Alternative approaches based on model-space sampling can
enable large-scale network inference, but their realization presents many
challenges. In this paper, we present new computational methods that make
large-scale nonlinear network inference tractable. First, we exploit the
topology of networks describing potential interactions among chemical species
to design improved "between-model" proposals for reversible-jump Markov chain
Monte Carlo. Second, we introduce a sensitivity-based determination of move
types which, when combined with network-aware proposals, yields significant
additional gains in sampling performance. These algorithms are demonstrated on
inference problems drawn from systems biology, with nonlinear differential
equation models of species interactions.
-
The step of expert taxa recognition currently slows down the response time of
many bioassessments. Shifting to quicker and cheaper state-of-the-art machine
learning approaches is still met with expert scepticism towards the ability and
logic of machines. In our study, we investigate both the differences in
accuracy and in the identification logic of taxonomic experts and machines. We
propose a systematic approach utilizing deep Convolutional Neural Nets with the
transfer learning paradigm and extensively evaluate it over a multi-pose
taxonomic dataset with hierarchical labels specifically created for this
comparison. We also study the prediction accuracy on different ranks of
taxonomic hierarchy in detail. Our results revealed that human experts using
actual specimens yield the lowest classification error ($\overline{CE}=6.1\%$).
However, a much faster, automated approach using deep Convolutional Neural Nets
comes close to human accuracy ($\overline{CE}=11.4\%$). Contrary to previous
findings in the literature, we find that for machines following a typical flat
classification approach commonly used in machine learning performs better than
forcing machines to adopt a hierarchical, local per parent node approach used
by human taxonomic experts. Finally, we publicly share our unique dataset to
serve as a public benchmark dataset in this field.
-
The advent of high--throughput transcription profiling technologies has
enabled identification of genes and pathways associated with disease, providing
new avenues for precision medicine. A key challenge is to analyze this data in
the context of the regulatory networks and pathways that control cellular
processes, while still obtaining insights that can be used to design new
diagnostic and therapeutic interventions. While classical differential
expression analysis provides specific and hence targetable gene-level insights,
it does not include any systems-level information. On the other hand, pathway
analyses integrate systems-level information with expression data, but are
often limited in their ability to indicate specific molecular targets. We
introduce GeneSurrounder, an analysis method that takes into account the
complex structure of interaction networks to identify specific genes that
disrupt pathway activity in a disease-specific manner. GeneSurrounder
integrates transcriptomic data and pathway network information in a novel
two-step procedure to detect genes that (i) appear to influence the expression
of other genes local to it in the network and (ii) are part of a subnetwork of
differentially expressed genes. Combined, this evidence can be used to pinpoint
specific genes that have a mechanistic role in the phenotype of interest.
Applying GeneSurrounder to three distinct ovarian cancer studies using a global
KEGG network, we show that our method is able to identify biologically relevant
genes and genes missed by single-gene association tests, integrate pathway and
expression data, and yield more consistent results across multiple studies of
the same phenotype than competing methods.
-
Meaningful laws of nature must be independent of the units employed to
measure the variables. The principle of similitude (Rayleigh 1915) or
dimensional homogeneity, states that only commensurable quantities (ones having
the same dimension) may be compared, therefore, meaningful laws of nature must
be homogeneous equations in their various units of measurement, a result which
was formalized in the $\rm \Pi$ theorem (Vaschy 1892; Buckingham 1914).
However, most relations in allometry do not satisfy this basic requirement,
including the `3/4 Law' (Kleiber 1932) that relates the basal metabolic rate
and body mass, which it is sometimes claimed to be the most fundamental
biological rate (Brown et al. 2004) and the closest to a law in life sciences
(West \& Brown 2004). Using the $\rm \Pi$ theorem, here we show that it is
possible to construct a unique homogeneous equation for the metabolic rates, in
agreement with data in the literature. We find that the variations in the
dependence of the metabolic rates on body mass are secondary, coming from
variations in the allometric dependence of the heart frequencies. This includes
not only different classes of animals (mammals, birds, invertebrates) but also
different exercise conditions (basal and maximal). Our results demonstrate that
most of the differences found in the allometric exponents (White et al. 2007)
are due to compare incommensurable quantities and that our dimensionally
homogenous formula, unify these differences into a single formulation. We
discuss the ecological implications of this new formulation in the context of
the Malthusian's, Fenchel's and the total energy consumed in a lifespan
relations.
-
There is a growing awareness that catastrophic phenomena in biology and
medicine can be mathematically represented in terms of saddle-node
bifurcations. In particular, the term `tipping', or critical transition has in
recent years entered the discourse of the general public in relation to
ecology, medicine, and public health. The saddle-node bifurcation and its
associated theory of catastrophe as put forth by Thom and Zeeman has seen
applications in a wide range of fields including molecular biophysics,
mesoscopic physics, and climate science. In this paper, we investigate a simple
model of a non-autonomous system with a time-dependent parameter $p(\tau)$ and
its corresponding `dynamic' (time-dependent) saddle-node bifurcation by the
modern theory of non-autonomous dynamical systems. We show that the actual
point of no return for a system undergoing tipping can be significantly delayed
in comparison to the {\em breaking time} $\hat{\tau}$ at which the
corresponding autonomous system with a time-independent parameter $p_{a}=
p(\hat{\tau})$ undergoes a bifurcation. A dimensionless parameter
$\alpha=\lambda p_0^3V^{-2}$ is introduced, in which $\lambda$ is the curvature
of the autonomous saddle-node bifurcation according to parameter $p(\tau)$,
which has an initial value of $p_{0}$ and a constant rate of change $V$. We
find that the breaking time $\hat{\tau}$ is always less than the actual point
of no return $\tau^*$ after which the critical transition is irreversible;
specifically, the relation $\tau^*-\hat{\tau}\simeq 2.338(\lambda
V)^{-\frac{1}{3}}$ is analytically obtained. For a system with a small $\lambda
V$, there exists a significant window of opportunity $(\hat{\tau},\tau^*)$
during which rapid reversal of the environment can save the system from
catastrophe.
-
In this paper we investigate the complexity of model selection and model
testing for dynamical systems with toric steady states. Such systems frequently
arise in the study of chemical reaction networks. We do this by formulating
these tasks as a constrained optimization problem in Euclidean space. This
optimization problem is known as a Euclidean distance problem; the complexity
of solving this problem is measured by an invariant called the Euclidean
distance (ED) degree. We determine closed-form expressions for the ED degree of
the steady states of several families of chemical reaction networks with toric
steady states and arbitrarily many reactions. To illustrate the utility of this
work we show how the ED degree can be used as a tool for estimating the
computational cost of solving the model testing and model selection problems.
-
The biomechanics of the human body gives subjects a high degree of freedom in
how they can execute movement. Nevertheless, subjects exhibit regularity in
their movement patterns. One way to account for this regularity is to suppose
that subjects select movement trajectories that are optimal in some sense. We
adopt the principle that human movements are optimal and develop a general
model for human movement patters that uses variational methods in the form of
optimal control theory to calculate trajectories of movement trajectories of
the body. We find that in this approach a constant of the motion that arises
from the model and which plays a role in the optimal control model that is
analogous to the role that the mechanical energy plays in classical physics. We
illustrate how this approach works in practice by using it to develop a model
of walking gait, making all the derivations and calculations in detail. We
finally show that this optimal control model of walking gait recovers in an
appropriate limit an existing model of walking gait which has been shown to
provide good estimates of many observed characteristics of walking gait.
-
Human movements are physical processes combining the classical mechanics of
the human body moving in space and the biomechanics of the muscles generating
the forces acting on the body under sophisticated sensory-motor control. The
characterization of the performance of human movements is a problem with
important applications in clinical and sports research. One way to characterize
movement performance is through measures of energy efficiency that relate the
mechanical energy of the body and metabolic energy expended by the muscles.
Such a characterization provides information about the performance of a
movement insofar as subjects select movements with the aim of maximizing the
energy efficiency. We examine the case of the energy efficiency of asynchronous
arm-cranking doing external mechanical work, that is, using the arms to turn an
asynchronous arm-crank that performs external mechanical work. We construct a
metabolic energy model and use it estimate how cranking may be performed with
maximum energy efficiency, and recover the intuitive result that for larger
external forces the crank-handles should be placed as far from the center of
the crank as is comfortable for the subject to turn. We further examine
mechanical advantage in asynchronous arm-cranking by constructing an idealized
system that is driven by a crank and which involves an adjustable mechanical
advantage, and analyze the case in which the avg. frequency is fixed and derive
the mechanical advantages that maximize energy efficiency.
-
Human movements are physical processes combining the classical mechanics of
the human body moving in space and the biomechanics of the muscles generating
the forces acting on the body under sophisticated sensory-motor control. One
way to characterize movement performance is through measures of energy
efficiency that relate the mechanical energy of the body and metabolic energy
expended by the muscles. We expect the practical utility of such measures to be
greater when human subjects execute movements that maximize energy efficiency.
We therefore seek to understand if and when subjects select movements with that
maximizing energy efficiency. We proceed using a model-based approach to
describe movements which perform a task requiring the body to add or remove
external mechanical work to or from an object. We use the specific example of
walking gaits doing external mechanical work by pulling a cart, and estimate
the relationship between the avg. walking speed and avg. step length. In the
limit where no external work is done, we find that the estimated maximum energy
efficiency walking gait is much slower than the walking gaits healthy adults
typically select. We then modify the situation of the walking gait by
introducing an idealized mechanical device that creates an adjustable
mechanical advantage. The walking gaits that maximize the energy efficiency
using the optimal mechanical advantage are again much slower than the walking
gaits healthy adults typically select. We finally modify the situation so that
the avg. walking speed is fixed and derive the pattern of the avg. step length
and mechanical advantage that maximize energy efficiency.
-
The biomechanics of the human body allow humans a range of possible ways of
executing movements to attain specific goals. This range of movement is limited
by a number of mechanical, biomechanical, or cognitive constraints. Shifts in
these limits result in changes available possible movements from which a
subject can select and can affect which movements a subject selects. Therefore
by understanding the limits on the range of movement we can come to a better
understanding of declines in movement performance due to disease or aging. In
this project, we look at how models for the limits on the range of movement can
be derived in a principled manner from a model of the movement. Using the
example of normal walking gaits, we develop a lower limit on the avg. walking
speed by examining the process by which the body restores mechanical energy
lost during walking, and we develop an upper limit on the avg. step length by
examining the forces the body can exert doing external mechanical work, in this
case, pulling a cart. Making slight changes to the model for normal walking
gaits, we develop a model of very slow walking gaits with avg. walking speeds
below the lower limit on normal walking gaits but that also has a lower limit
on the avg. walking speed. We note that the lowest avg. walking speeds observed
clinically fall into the range of very slow walking gaits so defined, and argue
that forms of bipedal locomotion with still lower speeds should be considered
distinct from walking gaits.
-
The biomechanics of the human body allow humans a range of possible ways of
executing movements to attain specific goals. Nevertheless, humans exhibit
significant patterns in how they execute movements. We propose that the
observed patterns of human movement arise because subjects select those ways to
execute movements that are, in a rigorous sense, optimal. In this project, we
show how this proposition can guide the development of computational models of
movement selection and thereby account for human movement patterns. We proceed
by first developing a movement utility formalism that operationalizes the
concept of a best or optimal way of executing a movement using a utility
function so that the problem of movement selection becomes the problem of
finding the movement that maximizes the utility function. Since the movement
utility formalism includes a contribution of the metabolic energy of the
movement (maximum utility movements try to minimize metabolic energy), we also
develop a metabolic energy formalism that we can use to construct estimators of
the metabolic energies of particular movements. We then show how we can
construct an estimator for the metabolic energies of normal walking gaits and
we use that estimator to construct a movement utility model of the selection of
normal walking gaits and show that the relationship between avg. walking speed
and avg. step length predicted by this model agrees with observation. We
conclude by proposing a physical mechanism that a subject might use to estimate
the metabolic energy of a movement in practice.
-
While the use of technology to provide accurate and objective measurements of
human movement performance is presently an area of great interest, efforts to
quantify the performance of movement are hampered by the lack of a principled
model that describes how a subject goes about making a movement. We put forward
a principled mathematical formalism that describes human movements using an
optimal control model in which the subject controls the jerk of the movement.
We construct the formalism by assuming that the movement a subject chooses to
make is better than the alternatives. We quantify the relative quality of
movements mathematically by specifying a cost functional that assigns a
numerical value to every possible movement; the subject makes the movement that
minimizes the cost functional. We develop the mathematical structure of
movements that minimize a cost functional, and observe that this development
parallels the development of analytical mechanics from the Principle of Least
Action. We derive a constant of the motion for human movements that plays a
role that is analogous to the role that the energy plays in classical
mechanics. We apply the formalism to the description of two movements: (1)
rapid, targeted movements of a computer mouse, and (2) finger-tapping, and show
that the constant of the motion that we have derived provides a useful value
with which we can characterize the performance of the movements. In the case of
rapid, targeted movements of a computer mouse, we show how the model of human
movement that we have developed can be made to agree with Fitts' law, and we
show how Fitts' law is related to the constant of the motion that we have
derived. We finally show that solutions exist within the model of human
movements that exhibit an oscillatory character reminiscent of tremor.
-
The lasso and elastic net linear regression models impose a
double-exponential prior distribution on the model parameters to achieve
regression shrinkage and variable selection, allowing the inference of robust
models from large data sets. However, there has been limited success in
deriving estimates for the full posterior distribution of regression
coefficients in these models, due to a need to evaluate analytically
intractable partition function integrals. Here, the Fourier transform is used
to express these integrals as complex-valued oscillatory integrals over
"regression frequencies". This results in an analytic expansion and stationary
phase approximation for the partition functions of the Bayesian lasso and
elastic net, where the non-differentiability of the double-exponential prior
has so far eluded such an approach. Use of this approximation leads to highly
accurate numerical estimates for the expectation values and marginal posterior
distributions of the regression coefficients, and allows for Bayesian inference
of much higher dimensional models than previously possible.
-
In this work, we consider the problem of estimating summary statistics to
characterise biochemical reaction networks of interest. Such networks are often
described using the framework of the Chemical Master Equation (CME). For
physically-realistic models, the CME is widely considered to be analytically
intractable. A variety of Monte Carlo algorithms have therefore been developed
to explore the dynamics of such networks empirically. Amongst them is the
multi-level method, which uses estimates from multiple ensembles of sample
paths of different accuracies to estimate a summary statistic of interest. {In
this work, we develop the multi-level method in two directions: (1) to increase
the robustness, reliability and performance of the multi-level method, we
implement an improved variance reduction method for generating the sample paths
of each ensemble; and (2) to improve computational performance, we demonstrate
the successful use of a different mechanism for choosing which ensembles should
be included in the multi-level algorithm.
-
We present a continuous model for structural brain connectivity based on the
Poisson point process. The model treats each streamline curve in a tractography
as an observed event in connectome space, here a product space of cortical
white matter boundaries. We approximate the model parameter via kernel density
estimation. To deal with the heavy computational burden, we develop a fast
parameter estimation method by pre-computing associated Legendre products of
the data, leveraging properties of the spherical heat kernel. We show how our
approach can be used to assess the quality of cortical parcellations with
respect to connectivty. We further present empirical results that suggest the
discrete connectomes derived from our model have substantially higher
test-retest reliability compared to standard methods.
-
Light microscopy as well as image acquisition and processing suffer from
physical and technical prejudices which preclude a correct interpretation of
biological observations which can be reflected in, e.g., medical and
pharmacological praxis. Using the examples of a diffracting microbead and
fluorescently labelled tissue, this article clarifies some ignored aspects of
image build-up in the light microscope and introduce algorithms for maximal
extraction of information from the 3D microscopic experiments. We provided a
correct set-up of the microscope and we sought a voxel (3D pixel) called an
electromagnetic centroid which localizes the information about the object. In
diffraction imaging and light emission, this voxel shows a minimal intensity
change in two consecutive optical cuts. This approach further enabled us to
identify z-stack of a DAPI-stained tissue section where at least one object of
a relevant fluorescent marker was in focus. The spatial corrections (overlaps)
of the DAPI-labelled region with in-focus autofluorescent regions then enabled
us to co-localize these three regions in the optimal way when considering
physical laws and information theory. We demonstrate that superresolution down
to the Nobelish level can be obtained from commonplace widefield bright-field
and fluorescence microscopy and bring new perspectives on co-localization in
fluorescent microscopy.
-
The color sensation evoked by an object depends on both the spectral power
distribution of the illumination and the reflectance properties of the object
being illuminated. The color sensation can be characterized by three
color-space values, such as XYZ, RGB, HSV, L*a*b*, etc. It is straightforward
to compute the three values given the illuminant and reflectance curves. The
converse process of computing a reflectance curve given the color-space values
and the illuminant is complicated by the fact that an infinite number of
different reflectance curves can give rise to a single set of color-space
values (metamerism). This paper presents five algorithms for generating a
reflectance curve from a specified sRGB triplet, written for a general
audience. The algorithms are designed to generate reflectance curves that are
similar to those found with naturally occurring colored objects. The computed
reflectance curves are compared to a database of thousands of reflectance
curves measured from paints and pigments available both commercially and in
nature, and the similarity is quantified. One particularly useful application
of these algorithms is in the field of computer graphics, where modeling color
transformations sometimes requires wavelength-specific information, such as
when modeling subtractive color mixture.
-
Biologists have long sought a way to explain how statistical properties of
genetic sequences emerged and are maintained through evolution. On the one
hand, non-random structures at different scales indicate a complex genome
organisation. On the other hand, single-strand symmetry has been scrutinised
using neutral models in which correlations are not considered or irrelevant,
contrary to empirical evidence. Different studies investigated these two
statistical features separately, reaching minimal consensus despite sustained
efforts. Here we unravel previously unknown symmetries in genetic sequences,
which are organized hierarchically through scales in which non-random
structures are known to be present. These observations are confirmed through
the statistical analysis of the human genome and explained through a simple
domain model. These results suggest that domain models which account for the
cumulative action of mobile elements can explain simultaneously non-random
structures and symmetries in genetic sequences.
-
We introduce and study a set of training-free methods of
information-theoretic and algorithmic complexity nature applied to DNA
sequences to identify their potential capabilities to determine nucleosomal
binding sites. We test our measures on well-studied genomic sequences of
different sizes drawn from different sources. The measures reveal the known in
vivo versus in vitro predictive discrepancies and uncover their potential to
pinpoint (high) nucleosome occupancy. We explore different possible signals
within and beyond the nucleosome length and find that complexity indices are
informative of nucleosome occupancy. We compare against the gold standard
(Kaplan model) and find similar and complementary results with the main
difference that our sequence complexity approach. For example, for high
occupancy, complexity-based scores outperform the Kaplan model for predicting
binding representing a significant advancement in predicting the highest
nucleosome occupancy following a training-free approach.





 Due to the scarcity of quantitative details about biological phenomena, quantitative modeling in systems biology can be compromised, especially at the subcellular scale. One way to get around this is qualitative modeling because it requires few to no quantitative information. One of the most popular qualitative modeling approaches is the Boolean network formalism. However, Boolean models allow variables to take only two values, which can be too simplistic in some cases. The present work proposes a modeling approach derived from Boolean networks where continuous logical operators are used and where edges can be tuned. Using continuous logical operators allows variables to be more finely valued while remaining qualitative. To consider that some biological interactions can be slower or weaker than other ones, edge states are also computed in order to modulate in speed and strength the signal they convey. The proposed formalism is illustrated on a toy network coming from the epidermal growth factor receptor signaling pathway. The obtained simulations show that continuous results are produced, thus allowing finer analysis. The simulations also show that modulating the signal conveyed by the edges allows to incorporate knowledge about the interactions they model. The goal is to provide enhancements in the ability of qualitative models to simulate the dynamics of biological networks while limiting the need of quantitative information.
Due to the scarcity of quantitative details about biological phenomena, quantitative modeling in systems biology can be compromised, especially at the subcellular scale. One way to get around this is qualitative modeling because it requires few to no quantitative information. One of the most popular qualitative modeling approaches is the Boolean network formalism. However, Boolean models allow variables to take only two values, which can be too simplistic in some cases. The present work proposes a modeling approach derived from Boolean networks where continuous logical operators are used and where edges can be tuned. Using continuous logical operators allows variables to be more finely valued while remaining qualitative. To consider that some biological interactions can be slower or weaker than other ones, edge states are also computed in order to modulate in speed and strength the signal they convey. The proposed formalism is illustrated on a toy network coming from the epidermal growth factor receptor signaling pathway. The obtained simulations show that continuous results are produced, thus allowing finer analysis. The simulations also show that modulating the signal conveyed by the edges allows to incorporate knowledge about the interactions they model. The goal is to provide enhancements in the ability of qualitative models to simulate the dynamics of biological networks while limiting the need of quantitative information.




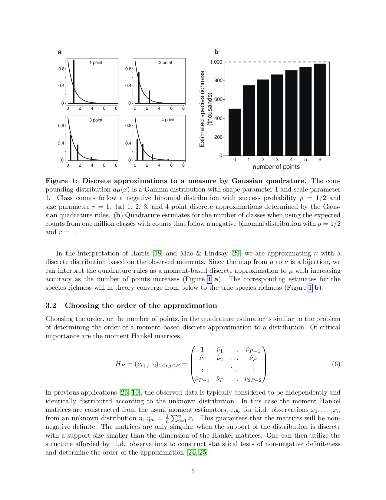 Estimation of the number of species or unobserved classes from a random sample of the underlying population is a ubiquitous problem in statistics. In classical settings, the size of the sample is usually small. New technologies such as high-throughput DNA sequencing have allowed for the sampling of extremely large and heterogeneous populations at scales not previously attainable or even considered. New algorithms are required that take advantage of the size of the data to account for heterogeneity, but are also sufficiently fast and scale well with large data. We present a non-parametric moment-based estimator that is both computationally efficient and is sufficiently flexible to account for heterogeneity in the abundances of underlying population. This estimator is based on an extension of a popular moment-based lower bound (Chao, 1984), originally developed by Harris (1959) but unattainable due to the lack of economical algorithms to solve the system of nonlinear equation required for estimation. We apply results from the classical moment problem to show that solutions can be obtained efficiently, allowing for estimators that are simultaneously conservative and use more information. This is critical for modern genomic applications, where there may be many large experiments that require the application of species estimation. We present applications of our estimator to estimating T-Cell receptor repertoire and dropout in single cell RNA-seq experiments.
Estimation of the number of species or unobserved classes from a random sample of the underlying population is a ubiquitous problem in statistics. In classical settings, the size of the sample is usually small. New technologies such as high-throughput DNA sequencing have allowed for the sampling of extremely large and heterogeneous populations at scales not previously attainable or even considered. New algorithms are required that take advantage of the size of the data to account for heterogeneity, but are also sufficiently fast and scale well with large data. We present a non-parametric moment-based estimator that is both computationally efficient and is sufficiently flexible to account for heterogeneity in the abundances of underlying population. This estimator is based on an extension of a popular moment-based lower bound (Chao, 1984), originally developed by Harris (1959) but unattainable due to the lack of economical algorithms to solve the system of nonlinear equation required for estimation. We apply results from the classical moment problem to show that solutions can be obtained efficiently, allowing for estimators that are simultaneously conservative and use more information. This is critical for modern genomic applications, where there may be many large experiments that require the application of species estimation. We present applications of our estimator to estimating T-Cell receptor repertoire and dropout in single cell RNA-seq experiments.




 Cryo-electron microscopy provides 2-D projection images of the 3-D electron scattering intensity of many instances of the particle under study (e.g., a virus). Both symmetry (rotational point groups) and heterogeneity are important aspects of biological particles and both aspects can be combined by describing the electron scattering intensity of the particle as a stochastic process with a symmetric probability law and therefore symmetric moments. A maximum likelihood estimator implemented by an expectation-maximization algorithm is described which estimates the unknown statistics of the electron scattering intensity stochastic process from images of instances of the particle. The algorithm is demonstrated on the bacteriophage HK97 and the virus N$\omega$V. The results are contrasted with existing algorithms which assume that each instance of the particle has the symmetry rather than the less restrictive assumption that the probability law has the symmetry.
Cryo-electron microscopy provides 2-D projection images of the 3-D electron scattering intensity of many instances of the particle under study (e.g., a virus). Both symmetry (rotational point groups) and heterogeneity are important aspects of biological particles and both aspects can be combined by describing the electron scattering intensity of the particle as a stochastic process with a symmetric probability law and therefore symmetric moments. A maximum likelihood estimator implemented by an expectation-maximization algorithm is described which estimates the unknown statistics of the electron scattering intensity stochastic process from images of instances of the particle. The algorithm is demonstrated on the bacteriophage HK97 and the virus N$\omega$V. The results are contrasted with existing algorithms which assume that each instance of the particle has the symmetry rather than the less restrictive assumption that the probability law has the symmetry.
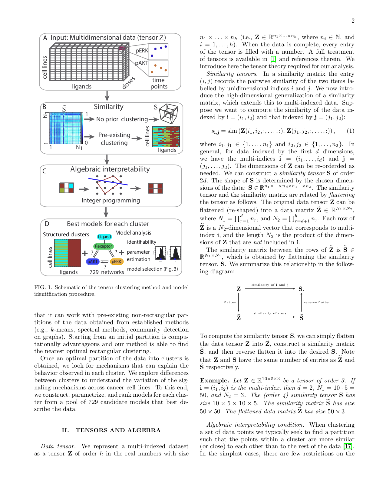



 We introduce a tensor-based clustering method to extract sparse, low-dimensional structure from high-dimensional, multi-indexed datasets. This framework is designed to enable detection of clusters of data in the presence of structural requirements which we encode as algebraic constraints in a linear program. Our clustering method is general and can be tailored to a variety of applications in science and industry. We illustrate our method on a collection of experiments measuring the response of genetically diverse breast cancer cell lines to an array of ligands. Each experiment consists of a cell line-ligand combination, and contains time-course measurements of the early-signalling kinases MAPK and AKT at two different ligand dose levels. By imposing appropriate structural constraints and respecting the multi-indexed structure of the data, the analysis of clusters can be optimized for biological interpretation and therapeutic understanding. We then perform a systematic, large-scale exploration of mechanistic models of MAPK-AKT crosstalk for each cluster. This analysis allows us to quantify the heterogeneity of breast cancer cell subtypes, and leads to hypotheses about the signalling mechanisms that mediate the response of the cell lines to ligands.
We introduce a tensor-based clustering method to extract sparse, low-dimensional structure from high-dimensional, multi-indexed datasets. This framework is designed to enable detection of clusters of data in the presence of structural requirements which we encode as algebraic constraints in a linear program. Our clustering method is general and can be tailored to a variety of applications in science and industry. We illustrate our method on a collection of experiments measuring the response of genetically diverse breast cancer cell lines to an array of ligands. Each experiment consists of a cell line-ligand combination, and contains time-course measurements of the early-signalling kinases MAPK and AKT at two different ligand dose levels. By imposing appropriate structural constraints and respecting the multi-indexed structure of the data, the analysis of clusters can be optimized for biological interpretation and therapeutic understanding. We then perform a systematic, large-scale exploration of mechanistic models of MAPK-AKT crosstalk for each cluster. This analysis allows us to quantify the heterogeneity of breast cancer cell subtypes, and leads to hypotheses about the signalling mechanisms that mediate the response of the cell lines to ligands.




 Pathway Tools is a bioinformatics software environment with a broad set of capabilities. The software provides genome-informatics tools such as a genome browser, sequence alignments, a genome-variant analyzer, and comparative-genomics operations. It offers metabolic-informatics tools, such as metabolic reconstruction, quantitative metabolic modeling, prediction of reaction atom mappings, and metabolic route search. Pathway Tools also provides regulatory-informatics tools, such as the ability to represent and visualize a wide range of regulatory interactions. The software creates and manages a type of organism-specific database called a Pathway/Genome Database (PGDB), which the software enables database curators to interactively edit. It supports web publishing of PGDBs and provides a large number of query, visualization, and omics-data analysis tools. Scientists around the world have created more than 9,800 PGDBs by using Pathway Tools, many of which are curated databases for important model organisms. Those PGDBs can be exchanged using a peer-to-peer database-sharing system called the PGDB Registry.
Pathway Tools is a bioinformatics software environment with a broad set of capabilities. The software provides genome-informatics tools such as a genome browser, sequence alignments, a genome-variant analyzer, and comparative-genomics operations. It offers metabolic-informatics tools, such as metabolic reconstruction, quantitative metabolic modeling, prediction of reaction atom mappings, and metabolic route search. Pathway Tools also provides regulatory-informatics tools, such as the ability to represent and visualize a wide range of regulatory interactions. The software creates and manages a type of organism-specific database called a Pathway/Genome Database (PGDB), which the software enables database curators to interactively edit. It supports web publishing of PGDBs and provides a large number of query, visualization, and omics-data analysis tools. Scientists around the world have created more than 9,800 PGDBs by using Pathway Tools, many of which are curated databases for important model organisms. Those PGDBs can be exchanged using a peer-to-peer database-sharing system called the PGDB Registry.



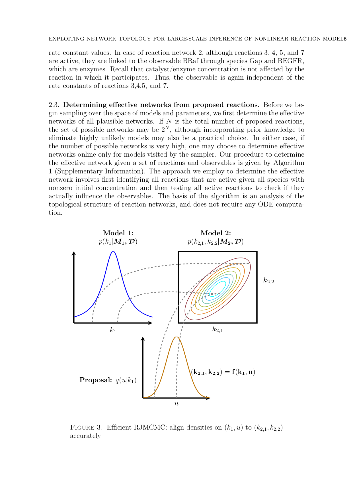
 The development of chemical reaction models aids understanding and prediction in areas ranging from biology to electrochemistry and combustion. A systematic approach to building reaction network models uses observational data not only to estimate unknown parameters, but also to learn model structure. Bayesian inference provides a natural approach to this data-driven construction of models. Yet traditional Bayesian model inference methodologies that numerically evaluate the evidence for each model are often infeasible for nonlinear reaction network inference, as the number of plausible models can be combinatorially large. Alternative approaches based on model-space sampling can enable large-scale network inference, but their realization presents many challenges. In this paper, we present new computational methods that make large-scale nonlinear network inference tractable. First, we exploit the topology of networks describing potential interactions among chemical species to design improved "between-model" proposals for reversible-jump Markov chain Monte Carlo. Second, we introduce a sensitivity-based determination of move types which, when combined with network-aware proposals, yields significant additional gains in sampling performance. These algorithms are demonstrated on inference problems drawn from systems biology, with nonlinear differential equation models of species interactions.
The development of chemical reaction models aids understanding and prediction in areas ranging from biology to electrochemistry and combustion. A systematic approach to building reaction network models uses observational data not only to estimate unknown parameters, but also to learn model structure. Bayesian inference provides a natural approach to this data-driven construction of models. Yet traditional Bayesian model inference methodologies that numerically evaluate the evidence for each model are often infeasible for nonlinear reaction network inference, as the number of plausible models can be combinatorially large. Alternative approaches based on model-space sampling can enable large-scale network inference, but their realization presents many challenges. In this paper, we present new computational methods that make large-scale nonlinear network inference tractable. First, we exploit the topology of networks describing potential interactions among chemical species to design improved "between-model" proposals for reversible-jump Markov chain Monte Carlo. Second, we introduce a sensitivity-based determination of move types which, when combined with network-aware proposals, yields significant additional gains in sampling performance. These algorithms are demonstrated on inference problems drawn from systems biology, with nonlinear differential equation models of species interactions.




 The step of expert taxa recognition currently slows down the response time of many bioassessments. Shifting to quicker and cheaper state-of-the-art machine learning approaches is still met with expert scepticism towards the ability and logic of machines. In our study, we investigate both the differences in accuracy and in the identification logic of taxonomic experts and machines. We propose a systematic approach utilizing deep Convolutional Neural Nets with the transfer learning paradigm and extensively evaluate it over a multi-pose taxonomic dataset with hierarchical labels specifically created for this comparison. We also study the prediction accuracy on different ranks of taxonomic hierarchy in detail. Our results revealed that human experts using actual specimens yield the lowest classification error ($\overline{CE}=6.1\%$). However, a much faster, automated approach using deep Convolutional Neural Nets comes close to human accuracy ($\overline{CE}=11.4\%$). Contrary to previous findings in the literature, we find that for machines following a typical flat classification approach commonly used in machine learning performs better than forcing machines to adopt a hierarchical, local per parent node approach used by human taxonomic experts. Finally, we publicly share our unique dataset to serve as a public benchmark dataset in this field.
The step of expert taxa recognition currently slows down the response time of many bioassessments. Shifting to quicker and cheaper state-of-the-art machine learning approaches is still met with expert scepticism towards the ability and logic of machines. In our study, we investigate both the differences in accuracy and in the identification logic of taxonomic experts and machines. We propose a systematic approach utilizing deep Convolutional Neural Nets with the transfer learning paradigm and extensively evaluate it over a multi-pose taxonomic dataset with hierarchical labels specifically created for this comparison. We also study the prediction accuracy on different ranks of taxonomic hierarchy in detail. Our results revealed that human experts using actual specimens yield the lowest classification error ($\overline{CE}=6.1\%$). However, a much faster, automated approach using deep Convolutional Neural Nets comes close to human accuracy ($\overline{CE}=11.4\%$). Contrary to previous findings in the literature, we find that for machines following a typical flat classification approach commonly used in machine learning performs better than forcing machines to adopt a hierarchical, local per parent node approach used by human taxonomic experts. Finally, we publicly share our unique dataset to serve as a public benchmark dataset in this field.




 The advent of high--throughput transcription profiling technologies has enabled identification of genes and pathways associated with disease, providing new avenues for precision medicine. A key challenge is to analyze this data in the context of the regulatory networks and pathways that control cellular processes, while still obtaining insights that can be used to design new diagnostic and therapeutic interventions. While classical differential expression analysis provides specific and hence targetable gene-level insights, it does not include any systems-level information. On the other hand, pathway analyses integrate systems-level information with expression data, but are often limited in their ability to indicate specific molecular targets. We introduce GeneSurrounder, an analysis method that takes into account the complex structure of interaction networks to identify specific genes that disrupt pathway activity in a disease-specific manner. GeneSurrounder integrates transcriptomic data and pathway network information in a novel two-step procedure to detect genes that (i) appear to influence the expression of other genes local to it in the network and (ii) are part of a subnetwork of differentially expressed genes. Combined, this evidence can be used to pinpoint specific genes that have a mechanistic role in the phenotype of interest. Applying GeneSurrounder to three distinct ovarian cancer studies using a global KEGG network, we show that our method is able to identify biologically relevant genes and genes missed by single-gene association tests, integrate pathway and expression data, and yield more consistent results across multiple studies of the same phenotype than competing methods.
The advent of high--throughput transcription profiling technologies has enabled identification of genes and pathways associated with disease, providing new avenues for precision medicine. A key challenge is to analyze this data in the context of the regulatory networks and pathways that control cellular processes, while still obtaining insights that can be used to design new diagnostic and therapeutic interventions. While classical differential expression analysis provides specific and hence targetable gene-level insights, it does not include any systems-level information. On the other hand, pathway analyses integrate systems-level information with expression data, but are often limited in their ability to indicate specific molecular targets. We introduce GeneSurrounder, an analysis method that takes into account the complex structure of interaction networks to identify specific genes that disrupt pathway activity in a disease-specific manner. GeneSurrounder integrates transcriptomic data and pathway network information in a novel two-step procedure to detect genes that (i) appear to influence the expression of other genes local to it in the network and (ii) are part of a subnetwork of differentially expressed genes. Combined, this evidence can be used to pinpoint specific genes that have a mechanistic role in the phenotype of interest. Applying GeneSurrounder to three distinct ovarian cancer studies using a global KEGG network, we show that our method is able to identify biologically relevant genes and genes missed by single-gene association tests, integrate pathway and expression data, and yield more consistent results across multiple studies of the same phenotype than competing methods.




 Meaningful laws of nature must be independent of the units employed to measure the variables. The principle of similitude (Rayleigh 1915) or dimensional homogeneity, states that only commensurable quantities (ones having the same dimension) may be compared, therefore, meaningful laws of nature must be homogeneous equations in their various units of measurement, a result which was formalized in the $\rm \Pi$ theorem (Vaschy 1892; Buckingham 1914). However, most relations in allometry do not satisfy this basic requirement, including the `3/4 Law' (Kleiber 1932) that relates the basal metabolic rate and body mass, which it is sometimes claimed to be the most fundamental biological rate (Brown et al. 2004) and the closest to a law in life sciences (West \& Brown 2004). Using the $\rm \Pi$ theorem, here we show that it is possible to construct a unique homogeneous equation for the metabolic rates, in agreement with data in the literature. We find that the variations in the dependence of the metabolic rates on body mass are secondary, coming from variations in the allometric dependence of the heart frequencies. This includes not only different classes of animals (mammals, birds, invertebrates) but also different exercise conditions (basal and maximal). Our results demonstrate that most of the differences found in the allometric exponents (White et al. 2007) are due to compare incommensurable quantities and that our dimensionally homogenous formula, unify these differences into a single formulation. We discuss the ecological implications of this new formulation in the context of the Malthusian's, Fenchel's and the total energy consumed in a lifespan relations.
Meaningful laws of nature must be independent of the units employed to measure the variables. The principle of similitude (Rayleigh 1915) or dimensional homogeneity, states that only commensurable quantities (ones having the same dimension) may be compared, therefore, meaningful laws of nature must be homogeneous equations in their various units of measurement, a result which was formalized in the $\rm \Pi$ theorem (Vaschy 1892; Buckingham 1914). However, most relations in allometry do not satisfy this basic requirement, including the `3/4 Law' (Kleiber 1932) that relates the basal metabolic rate and body mass, which it is sometimes claimed to be the most fundamental biological rate (Brown et al. 2004) and the closest to a law in life sciences (West \& Brown 2004). Using the $\rm \Pi$ theorem, here we show that it is possible to construct a unique homogeneous equation for the metabolic rates, in agreement with data in the literature. We find that the variations in the dependence of the metabolic rates on body mass are secondary, coming from variations in the allometric dependence of the heart frequencies. This includes not only different classes of animals (mammals, birds, invertebrates) but also different exercise conditions (basal and maximal). Our results demonstrate that most of the differences found in the allometric exponents (White et al. 2007) are due to compare incommensurable quantities and that our dimensionally homogenous formula, unify these differences into a single formulation. We discuss the ecological implications of this new formulation in the context of the Malthusian's, Fenchel's and the total energy consumed in a lifespan relations.




 There is a growing awareness that catastrophic phenomena in biology and medicine can be mathematically represented in terms of saddle-node bifurcations. In particular, the term `tipping', or critical transition has in recent years entered the discourse of the general public in relation to ecology, medicine, and public health. The saddle-node bifurcation and its associated theory of catastrophe as put forth by Thom and Zeeman has seen applications in a wide range of fields including molecular biophysics, mesoscopic physics, and climate science. In this paper, we investigate a simple model of a non-autonomous system with a time-dependent parameter $p(\tau)$ and its corresponding `dynamic' (time-dependent) saddle-node bifurcation by the modern theory of non-autonomous dynamical systems. We show that the actual point of no return for a system undergoing tipping can be significantly delayed in comparison to the {\em breaking time} $\hat{\tau}$ at which the corresponding autonomous system with a time-independent parameter $p_{a}= p(\hat{\tau})$ undergoes a bifurcation. A dimensionless parameter $\alpha=\lambda p_0^3V^{-2}$ is introduced, in which $\lambda$ is the curvature of the autonomous saddle-node bifurcation according to parameter $p(\tau)$, which has an initial value of $p_{0}$ and a constant rate of change $V$. We find that the breaking time $\hat{\tau}$ is always less than the actual point of no return $\tau^*$ after which the critical transition is irreversible; specifically, the relation $\tau^*-\hat{\tau}\simeq 2.338(\lambda V)^{-\frac{1}{3}}$ is analytically obtained. For a system with a small $\lambda V$, there exists a significant window of opportunity $(\hat{\tau},\tau^*)$ during which rapid reversal of the environment can save the system from catastrophe.
There is a growing awareness that catastrophic phenomena in biology and medicine can be mathematically represented in terms of saddle-node bifurcations. In particular, the term `tipping', or critical transition has in recent years entered the discourse of the general public in relation to ecology, medicine, and public health. The saddle-node bifurcation and its associated theory of catastrophe as put forth by Thom and Zeeman has seen applications in a wide range of fields including molecular biophysics, mesoscopic physics, and climate science. In this paper, we investigate a simple model of a non-autonomous system with a time-dependent parameter $p(\tau)$ and its corresponding `dynamic' (time-dependent) saddle-node bifurcation by the modern theory of non-autonomous dynamical systems. We show that the actual point of no return for a system undergoing tipping can be significantly delayed in comparison to the {\em breaking time} $\hat{\tau}$ at which the corresponding autonomous system with a time-independent parameter $p_{a}= p(\hat{\tau})$ undergoes a bifurcation. A dimensionless parameter $\alpha=\lambda p_0^3V^{-2}$ is introduced, in which $\lambda$ is the curvature of the autonomous saddle-node bifurcation according to parameter $p(\tau)$, which has an initial value of $p_{0}$ and a constant rate of change $V$. We find that the breaking time $\hat{\tau}$ is always less than the actual point of no return $\tau^*$ after which the critical transition is irreversible; specifically, the relation $\tau^*-\hat{\tau}\simeq 2.338(\lambda V)^{-\frac{1}{3}}$ is analytically obtained. For a system with a small $\lambda V$, there exists a significant window of opportunity $(\hat{\tau},\tau^*)$ during which rapid reversal of the environment can save the system from catastrophe.




 In this paper we investigate the complexity of model selection and model testing for dynamical systems with toric steady states. Such systems frequently arise in the study of chemical reaction networks. We do this by formulating these tasks as a constrained optimization problem in Euclidean space. This optimization problem is known as a Euclidean distance problem; the complexity of solving this problem is measured by an invariant called the Euclidean distance (ED) degree. We determine closed-form expressions for the ED degree of the steady states of several families of chemical reaction networks with toric steady states and arbitrarily many reactions. To illustrate the utility of this work we show how the ED degree can be used as a tool for estimating the computational cost of solving the model testing and model selection problems.
In this paper we investigate the complexity of model selection and model testing for dynamical systems with toric steady states. Such systems frequently arise in the study of chemical reaction networks. We do this by formulating these tasks as a constrained optimization problem in Euclidean space. This optimization problem is known as a Euclidean distance problem; the complexity of solving this problem is measured by an invariant called the Euclidean distance (ED) degree. We determine closed-form expressions for the ED degree of the steady states of several families of chemical reaction networks with toric steady states and arbitrarily many reactions. To illustrate the utility of this work we show how the ED degree can be used as a tool for estimating the computational cost of solving the model testing and model selection problems.




 In this work, we consider the problem of estimating summary statistics to characterise biochemical reaction networks of interest. Such networks are often described using the framework of the Chemical Master Equation (CME). For physically-realistic models, the CME is widely considered to be analytically intractable. A variety of Monte Carlo algorithms have therefore been developed to explore the dynamics of such networks empirically. Amongst them is the multi-level method, which uses estimates from multiple ensembles of sample paths of different accuracies to estimate a summary statistic of interest. {In this work, we develop the multi-level method in two directions: (1) to increase the robustness, reliability and performance of the multi-level method, we implement an improved variance reduction method for generating the sample paths of each ensemble; and (2) to improve computational performance, we demonstrate the successful use of a different mechanism for choosing which ensembles should be included in the multi-level algorithm.
In this work, we consider the problem of estimating summary statistics to characterise biochemical reaction networks of interest. Such networks are often described using the framework of the Chemical Master Equation (CME). For physically-realistic models, the CME is widely considered to be analytically intractable. A variety of Monte Carlo algorithms have therefore been developed to explore the dynamics of such networks empirically. Amongst them is the multi-level method, which uses estimates from multiple ensembles of sample paths of different accuracies to estimate a summary statistic of interest. {In this work, we develop the multi-level method in two directions: (1) to increase the robustness, reliability and performance of the multi-level method, we implement an improved variance reduction method for generating the sample paths of each ensemble; and (2) to improve computational performance, we demonstrate the successful use of a different mechanism for choosing which ensembles should be included in the multi-level algorithm.




 We present a continuous model for structural brain connectivity based on the Poisson point process. The model treats each streamline curve in a tractography as an observed event in connectome space, here a product space of cortical white matter boundaries. We approximate the model parameter via kernel density estimation. To deal with the heavy computational burden, we develop a fast parameter estimation method by pre-computing associated Legendre products of the data, leveraging properties of the spherical heat kernel. We show how our approach can be used to assess the quality of cortical parcellations with respect to connectivty. We further present empirical results that suggest the discrete connectomes derived from our model have substantially higher test-retest reliability compared to standard methods.
We present a continuous model for structural brain connectivity based on the Poisson point process. The model treats each streamline curve in a tractography as an observed event in connectome space, here a product space of cortical white matter boundaries. We approximate the model parameter via kernel density estimation. To deal with the heavy computational burden, we develop a fast parameter estimation method by pre-computing associated Legendre products of the data, leveraging properties of the spherical heat kernel. We show how our approach can be used to assess the quality of cortical parcellations with respect to connectivty. We further present empirical results that suggest the discrete connectomes derived from our model have substantially higher test-retest reliability compared to standard methods.



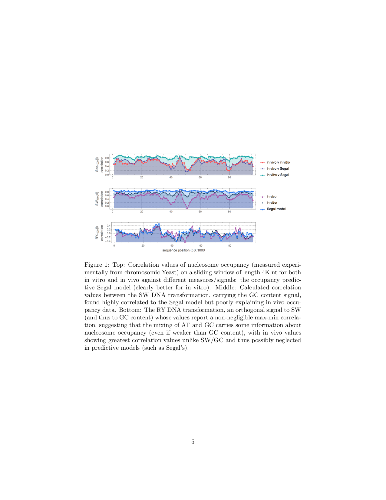
 We introduce and study a set of training-free methods of information-theoretic and algorithmic complexity nature applied to DNA sequences to identify their potential capabilities to determine nucleosomal binding sites. We test our measures on well-studied genomic sequences of different sizes drawn from different sources. The measures reveal the known in vivo versus in vitro predictive discrepancies and uncover their potential to pinpoint (high) nucleosome occupancy. We explore different possible signals within and beyond the nucleosome length and find that complexity indices are informative of nucleosome occupancy. We compare against the gold standard (Kaplan model) and find similar and complementary results with the main difference that our sequence complexity approach. For example, for high occupancy, complexity-based scores outperform the Kaplan model for predicting binding representing a significant advancement in predicting the highest nucleosome occupancy following a training-free approach.
We introduce and study a set of training-free methods of information-theoretic and algorithmic complexity nature applied to DNA sequences to identify their potential capabilities to determine nucleosomal binding sites. We test our measures on well-studied genomic sequences of different sizes drawn from different sources. The measures reveal the known in vivo versus in vitro predictive discrepancies and uncover their potential to pinpoint (high) nucleosome occupancy. We explore different possible signals within and beyond the nucleosome length and find that complexity indices are informative of nucleosome occupancy. We compare against the gold standard (Kaplan model) and find similar and complementary results with the main difference that our sequence complexity approach. For example, for high occupancy, complexity-based scores outperform the Kaplan model for predicting binding representing a significant advancement in predicting the highest nucleosome occupancy following a training-free approach.