-
Due to the scarcity of quantitative details about biological phenomena,
quantitative modeling in systems biology can be compromised, especially at the
subcellular scale. One way to get around this is qualitative modeling because
it requires few to no quantitative information. One of the most popular
qualitative modeling approaches is the Boolean network formalism. However,
Boolean models allow variables to take only two values, which can be too
simplistic in some cases. The present work proposes a modeling approach derived
from Boolean networks where continuous logical operators are used and where
edges can be tuned. Using continuous logical operators allows variables to be
more finely valued while remaining qualitative. To consider that some
biological interactions can be slower or weaker than other ones, edge states
are also computed in order to modulate in speed and strength the signal they
convey. The proposed formalism is illustrated on a toy network coming from the
epidermal growth factor receptor signaling pathway. The obtained simulations
show that continuous results are produced, thus allowing finer analysis. The
simulations also show that modulating the signal conveyed by the edges allows
to incorporate knowledge about the interactions they model. The goal is to
provide enhancements in the ability of qualitative models to simulate the
dynamics of biological networks while limiting the need of quantitative
information.
-
We describe and analyse Levenberg-Marquardt methods for solving systems of
nonlinear equations. More specifically, we propose an adaptive formula for the
Levenberg-Marquardt parameter and analyse the local convergence of the method
under H\"{o}lder metric subregularity of the function defining the equation and
H\"older continuity of its gradient mapping. Further, we analyse the local
convergence of the method under the additional assumption that the
\L{}ojasiewicz gradient inequality holds. We finally report encouraging
numerical results confirming the theoretical findings for the problem of
computing moiety conserved steady states in biochemical reaction networks. This
problem can be cast as finding a solution of a system of nonlinear equations,
where the associated mapping satisfies the \L{}ojasiewicz gradient inequality
assumption.
-
We introduce a tensor-based clustering method to extract sparse,
low-dimensional structure from high-dimensional, multi-indexed datasets. This
framework is designed to enable detection of clusters of data in the presence
of structural requirements which we encode as algebraic constraints in a linear
program. Our clustering method is general and can be tailored to a variety of
applications in science and industry. We illustrate our method on a collection
of experiments measuring the response of genetically diverse breast cancer cell
lines to an array of ligands. Each experiment consists of a cell line-ligand
combination, and contains time-course measurements of the early-signalling
kinases MAPK and AKT at two different ligand dose levels. By imposing
appropriate structural constraints and respecting the multi-indexed structure
of the data, the analysis of clusters can be optimized for biological
interpretation and therapeutic understanding. We then perform a systematic,
large-scale exploration of mechanistic models of MAPK-AKT crosstalk for each
cluster. This analysis allows us to quantify the heterogeneity of breast cancer
cell subtypes, and leads to hypotheses about the signalling mechanisms that
mediate the response of the cell lines to ligands.
-
We map a class of well-mixed stochastic models of biochemical feedback in
steady state to the mean-field Ising model near the critical point. The mapping
provides an effective temperature, magnetic field, order parameter, and heat
capacity that can be extracted from biological data without fitting or
knowledge of the underlying molecular details. We demonstrate this procedure on
fluorescence data from mouse T cells, which reveals distinctions between how
the cells respond to different drugs. We also show that the heat capacity
allows inference of absolute molecule number from fluorescence intensity. We
explain this result in terms of the underlying fluctuations and demonstrate the
generality of our work.
-
Temporal variations in biological systems and more generally in natural
sciences are typically modelled as a set of Ordinary, Partial, or Stochastic
Differential or Difference Equations. Algorithms for learning the structure and
the parameters of a dynamical system are distinguished based on whether time is
discrete or continuous, observations are time-series or time-course, and
whether the system is deterministic or stochastic, however, there is no
approach able to handle the various types of dynamical systems simultaneously.
In this paper, we present a unified approach to infer both the structure and
the parameters of nonlinear dynamical systems of any type under the restriction
of being linear with respect to the unknown parameters. Our approach, which is
named Unified Sparse Dynamics Learning (USDL), constitutes of two steps. First,
an atemporal system of equations is derived through the application of the weak
formulation. Then, assuming a sparse representation for the dynamical system,
we show that the inference problem can be expressed as a sparse signal recovery
problem, allowing the application of an extensive body of algorithms and
theoretical results. Results on simulated data demonstrate the efficacy and
superiority of the USDL algorithm as a function of the experimental setup
(sample size, number of time measurements, number of interventions, noise
level). Additionally, USDL's accuracy significantly correlates with theoretical
metrics such as the exact recovery coefficient. On real single-cell data, the
proposed approach is able to induce high-confidence subgraphs of the signaling
pathway. USDL algorithm has been integrated in SCENERY
(\url{http://scenery.csd.uoc.gr/}); an online tool for single-cell mass
cytometry analytics.
-
The advent of high--throughput transcription profiling technologies has
enabled identification of genes and pathways associated with disease, providing
new avenues for precision medicine. A key challenge is to analyze this data in
the context of the regulatory networks and pathways that control cellular
processes, while still obtaining insights that can be used to design new
diagnostic and therapeutic interventions. While classical differential
expression analysis provides specific and hence targetable gene-level insights,
it does not include any systems-level information. On the other hand, pathway
analyses integrate systems-level information with expression data, but are
often limited in their ability to indicate specific molecular targets. We
introduce GeneSurrounder, an analysis method that takes into account the
complex structure of interaction networks to identify specific genes that
disrupt pathway activity in a disease-specific manner. GeneSurrounder
integrates transcriptomic data and pathway network information in a novel
two-step procedure to detect genes that (i) appear to influence the expression
of other genes local to it in the network and (ii) are part of a subnetwork of
differentially expressed genes. Combined, this evidence can be used to pinpoint
specific genes that have a mechanistic role in the phenotype of interest.
Applying GeneSurrounder to three distinct ovarian cancer studies using a global
KEGG network, we show that our method is able to identify biologically relevant
genes and genes missed by single-gene association tests, integrate pathway and
expression data, and yield more consistent results across multiple studies of
the same phenotype than competing methods.
-
This paper proposes a control theoretic framework to model and analyze the
self-organized pattern formation of molecular concentrations in biomolecular
communication networks, emerging applications in synthetic biology. In
biomolecular communication networks, bionanomachines, or biological cells,
communicate with each other using a cell-to-cell communication mechanism
mediated by a diffusible signaling molecule, thereby the dynamics of molecular
concentrations are approximately modeled as a reaction-diffusion system with a
single diffuser. We first introduce a feedback model representation of the
reaction-diffusion system and provide a systematic local stability/instability
analysis tool using the root locus of the feedback system. The instability
analysis then allows us to analytically derive the conditions for the
self-organized spatial pattern formation, or Turing pattern formation, of the
bionanomachines. We propose a novel synthetic biocircuit motif called
activator-repressor-diffuser system and show that it is one of the minimum
biomolecular circuits that admit self-organized patterns over cell population.
-
In this paper we investigate the complexity of model selection and model
testing for dynamical systems with toric steady states. Such systems frequently
arise in the study of chemical reaction networks. We do this by formulating
these tasks as a constrained optimization problem in Euclidean space. This
optimization problem is known as a Euclidean distance problem; the complexity
of solving this problem is measured by an invariant called the Euclidean
distance (ED) degree. We determine closed-form expressions for the ED degree of
the steady states of several families of chemical reaction networks with toric
steady states and arbitrarily many reactions. To illustrate the utility of this
work we show how the ED degree can be used as a tool for estimating the
computational cost of solving the model testing and model selection problems.
-
Transcription factors (TFs) exert their regulatory action by binding to DNA
with specific sequence preferences. However, different TFs can partially share
their binding sequences due to their common evolutionary origin. This
`redundancy' of binding defines a way of organizing TFs in `motif families' by
grouping TFs with similar binding preferences. Since these ultimately define
the TF target genes, the motif family organization entails information about
the structure of transcriptional regulation as it has been shaped by evolution.
Focusing on the human TF repertoire, we show that a one-parameter evolutionary
model of the Birth-Death-Innovation type can explain the TF empirical
ripartition in motif families, and allows to highlight the relevant
evolutionary forces at the origin of this organization. Moreover, the model
allows to pinpoint few deviations from the neutral scenario it assumes: three
over-expanded families (including HOX and FOX genes), a set of `singleton' TFs
for which duplication seems to be selected against, and a higher-than-average
rate of diversification of the binding preferences of TFs with a Zinc Finger
DNA binding domain. Finally, a comparison of the TF motif family organization
in different eukaryotic species suggests an increase of redundancy of binding
with organism complexity.
-
Controlling stochastic reactions networks is a challenging problem with
important implications in various fields such as systems and synthetic biology.
Various regulation motifs have been discovered or posited over the recent
years, the most recent one being the so-called Antithetic Integral Control
(AIC) motif in Briat et al. (Cell Systems, 2016). Several favorable properties
for the AIC motif have been demonstrated for classes of reaction networks that
satisfy certain irreducibility, ergodicity and output controllability
conditions. Here we address the problem of verifying these conditions for large
sets of reaction networks with fixed topology using two different approaches.
The first one is quantitative and relies on the notion of interval matrices
while the second one is qualitative and is based on sign properties of
matrices. The obtained results lie in the same spirit as those obtained in
Briat et al. (Cell Systems, 2016) where properties of reaction networks are
independently characterized in terms of control theoretic concepts, linear
programming conditions and graph theoretic conditions.
-
The stochastic dynamics of biochemical networks are usually modelled with the
chemical master equation (CME). The stationary distributions of CMEs are seldom
solvable analytically, and numerical methods typically produce estimates with
uncontrolled errors. Here, we introduce mathematical programming approaches
that yield approximations of these distributions with computable error bounds
which enable the verification of their accuracy. First, we use semidefinite
programming to compute increasingly tighter upper and lower bounds on the
moments of the stationary distributions for networks with rational
propensities. Second, we use these moment bounds to formulate linear programs
that yield convergent upper and lower bounds on the stationary distributions
themselves, their marginals and stationary averages. The bounds obtained also
provide a computational test for the uniqueness of the distribution. In the
unique case, the bounds form an approximation of the stationary distribution
with a computable bound on its error. In the non-unique case, our approach
yields converging approximations of the ergodic distributions. We illustrate
our methodology through several biochemical examples taken from the literature:
Schl\"ogl's model for a chemical bifurcation, a two-dimensional toggle switch,
a model for bursty gene expression, and a dimerisation model with multiple
stationary distributions.
-
The formalism of partial information decomposition provides independent or
non-overlapping components constituting total information content provided by a
set of source variables about the target variable. These components are
recognised as unique information, synergistic information and, redundant
information. The metric of net synergy, conceived as the difference between
synergistic and redundant information, is capable of detecting synergy,
redundancy and, information independence among stochastic variables. And it can
be quantified, as it is done here, using appropriate combinations of different
Shannon mutual information terms. Utilisation of such a metric in network
motifs with the nodes representing different biochemical species, involved in
information sharing, uncovers rich store for interesting results. In the
current study, we make use of this formalism to obtain a comprehensive
understanding of the relative information processing mechanism in a diamond
motif and two of its sub-motifs namely bifurcation and integration motif
embedded within the diamond motif. The emerging patterns of synergy and
redundancy and their effective contribution towards ensuring high fidelity
information transmission are duly compared in the sub-motifs and independent
motifs (bifurcation and integration). In this context, the crucial roles played
by various time scales and activation coefficients in the network topologies
are especially emphasised. We show that the origin of synergy and redundancy in
information transmission can be physically justified by decomposing diamond
motif into bifurcation and integration motif.
-
Sensitivity analysis of biochemical reactions aims at quantifying the
dependence of the reaction dynamics on the reaction rates. The computation of
the parameter sensitivities, however, poses many computational challenges when
taking stochastic noise into account. This paper proposes a new finite
difference method for efficiently computing sensitivities of biochemical
reactions. We employ propensity bounds of reactions to couple the simulation of
the nominal and perturbed processes. The exactness of the simulation is
reserved by applying the rejection-based mechanism. For each simulation step,
the nominal and perturbed processes under our coupling strategy are
synchronized and often jump together, increasing their positive correlation and
hence reducing the variance of the estimator. The distinctive feature of our
approach in comparison with existing coupling approaches is that it only needs
to maintain a single data structure storing propensity bounds of reactions
during the simulation of the nominal and perturbed processes. Our approach
allows to computing sensitivities of many reaction rates simultaneously.
Moreover, the data structure does not require to be updated frequently, hence
improving the computational cost. This feature is especially useful when
applied to large reaction networks. We benchmark our method on biological
reaction models to prove its applicability and efficiency.
-
In the immune system, T cells can quickly discriminate between foreign and
self ligands with high accuracy. There is evidence that T-cells achieve this
remarkable performance utilizing a network architecture based on a
generalization of kinetic proofreading (KPR). KPR-based mechanisms actively
consume energy to increase the specificity beyond what is possible in
equilibrium.An important theoretical question that arises is to understand the
trade-offs and fundamental limits on accuracy, speed, and dissipation (energy
consumption) in KPR and its generalization. Here, we revisit this question
through numerical simulations where we simultaneously measure the speed,
accuracy, and energy consumption of the KPR and adaptive sorting networks for
different parameter choices. Our simulations highlight the existence of a
'feasible operating regime' in the speed-energy-accuracy plane where T-cells
can quickly differentiate between foreign and self ligands at reasonable energy
expenditure. We give general arguments for why we expect this feasible
operating regime to be a generic property of all KPR-based biochemical networks
and discuss implications for our understanding of the T cell receptor circuit.
-
Model-based prediction of stochastic noise in biomolecular reactions often
resorts to approximation with unknown precision. As a result, unexpected
stochastic fluctuation causes a headache for the designers of biomolecular
circuits. This paper proposes a convex optimization approach to quantifying the
steady state moments of molecular copy counts with theoretical rigor. We show
that the stochastic moments lie in a convex semi-algebraic set specified by
linear matrix inequalities. Thus, the upper and the lower bounds of some
moments can be computed by a semidefinite program. Using a protein dimerization
process as an example, we demonstrate that the proposed method can precisely
predict the mean and the variance of the copy number of the monomer protein.
-
Investigations of topological uniqueness of gene interaction networks in
cancer cells are essential for understanding this disease. Based on the random
matrix theory, we study the distribution of the nearest neighbor level spacings
$P(s)$ of interaction matrices for gene networks in human cancer cells. The
interaction matrices are computed using the Cancer Network Galaxy (TCNG)
database, which is a repository of gene interactions inferred by a Bayesian
network model. 256 NCBI GEO entries regarding gene expressions in human cancer
cells have been selected for the Bayesian network calculations in TCNG. We
observe the Wigner distribution of $P(s)$ when the gene networks are dense
networks that have more than $\sim 38,000$ edges. In the opposite case, when
the networks have smaller numbers of edges, the distribution $P(s)$ becomes the
Poisson distribution. We investigate relevance of $P(s)$ both to the size of
the networks and to edge frequencies that manifest reliance of the inferred
gene interactions.
-
Biological systems are driven by intricate interactions among the complex
array of molecules that comprise the cell. Many methods have been developed to
reconstruct network models of those interactions. These methods often draw on
large numbers of samples with measured gene expression profiles to infer
connections between genes (or gene products). The result is an aggregate
network model representing a single estimate for the likelihood of each
interaction, or "edge," in the network. While informative, aggregate models
fail to capture the heterogeneity that is represented in any population. Here
we propose a method to reverse engineer sample-specific networks from aggregate
network models. We demonstrate the accuracy and applicability of our approach
in several data sets, including simulated data, microarray expression data from
synchronized yeast cells, and RNA-seq data collected from human lymphoblastoid
cell lines. We show that these sample-specific networks can be used to study
changes in network topology across time and to characterize shifts in gene
regulation that may not be apparent in expression data. We believe the ability
to generate sample-specific networks will greatly facilitate the application of
network methods to the increasingly large, complex, and heterogeneous
multi-omic data sets that are currently being generated, and ultimately support
the emerging field of precision network medicine.
-
Phenotypical variability in the absence of genetic variation often reflects
complex energetic landscapes associated with underlying gene regulatory
networks (GRNs). In this view, different phenotypes are associated with
alternative states of complex nonlinear systems: stable attractors in
deterministic models or modes of stationary distributions in stochastic
descriptions. We provide theoretical and practical characterizations of these
landscapes, specifically focusing on stochastic slow promoter kinetics, a time
scale relevant when transcription factor binding and unbinding are affected by
epigenetic processes like DNA methylation and chromatin remodeling. In this
case, largely unexplored except for numerical simulations, adiabatic
approximations of promoter kinetics are not appropriate. In contrast to the
existing literature, we provide rigorous analytic characterizations of multiple
modes. A general formal approach gives insight into the influence of parameters
and the prediction of how changes in GRN wiring, for example through mutations
or artificial interventions, impact the possible number, location, and
likelihood of alternative states. We adapt tools from the mathematical field of
singular perturbation theory to represent stationary distributions of Chemical
Master Equations for GRNs as mixtures of Poisson distributions and obtain
explicit formulas for the locations and probabilities of metastable states as a
function of the parameters describing the system. As illustrations, the theory
is used to tease out the role of cooperative binding in stochastic models in
comparison to deterministic models, and applications are given to various model
systems, such as toggle switches in isolation or in communicating populations,
a synthetic oscillator and a trans-differentiation network.
-
We consider stochastically modeled reaction networks and prove that if a
constant solution to the Kolmogorov forward equation decays fast enough
relatively to the transition rates, then the model is non-explosive. In
particular, complex balanced reaction networks are non-explosive.
-
The graph-related symmetries of a reaction network give rise to certain
special equilibria (such as complex balanced equilibria) in deterministic
models of dynamics of the reaction network. Correspondingly, in the stochastic
setting, when modeled as a continuous-time Markov chain, these symmetries give
rise to certain special stationary measures. Previous work by Anderson, Craciun
and Kurtz identified stationary distributions of a complex balanced network;
later Cappelletti and Wiuf developed the notion of complex balancing for
stochastic systems. We define and establish the relations between reaction
balanced measure, complex balanced measure, reaction vector balanced measure,
and cycle balanced measure and prove that with mild additional hypotheses, the
former two are stationary distributions. Furthermore, in spirit of earlier work
by Joshi, we give sufficient conditions under which detailed balance of the
stationary distribution of Markov chain models implies the existence of
positive detailed balance equilibria for the related deterministic reaction
network model. Finally, we provide a complete map of the implications between
balancing properties of deterministic and corresponding stochastic reaction
systems, such as complex balance, reaction balance, reaction vector balance and
cycle balance.
-
To estimate the time, many organisms, ranging from cyanobacteria to animals,
employ a circadian clock which is based on a limit-cycle oscillator that can
tick autonomously with a nearly 24h period. Yet, a limit-cycle oscillator is
not essential for knowing the time, as exemplified by bacteria that possess an
'hourglass': a system that when forced by an oscillatory light input exhibits
robust oscillations from which the organism can infer the time, but that in the
absence of driving relaxes to a stable fixed point. Here, using models of the
Kai system of cyanobacteria, we compare a limit- cycle oscillator with two
hourglass models, one that without driving relaxes exponentially and one that
does so in an oscillatory fashion. In the limit of low input-noise, all three
systems are equally informative on time, yet in the regime of high input-noise
the limit-cycle oscillator is far superior. The same behavior is found in the
Stuart-Landau model, indicating that our result is universal.
-
Many biological activities are induced by cellular chemical reactions of
diffusing reactants. The dynamics of such systems can be captured by stochastic
reaction networks. A recent numerical study has shown that diffusion can
significantly enhance the fluctuations in gene regulatory networks. However,
the general relation between diffusion and stochastic system dynamics remains
unveiled. Here we examine the general relation and find the universal law under
which the steady-state distribution in complex balanced networks is
diffusion-independent. Here, complex balance is the non-equilibrium
generalization of detailed balance. We also find that for a diffusion-included
network with a Poisson-like steady-state distribution, the diffusion can be
ignored at steady-state. We then derive a necessary and sufficient condition
for such steady-state network distributions. Moreover, we exactly prove that
the stochastic dynamics of linear reaction networks are unaffected by diffusion
at any arbitrary time. Our findings shed light on the fundamental question of
when diffusion can be neglected, or (if non-negligible) its effects on the
stochastic dynamics of the reaction network.
-
Networks are ubiquitous in biology where they encode connectivity patterns at
all scales of organization, from molecular to the biome. However, biological
networks are noisy due to the limitations of the technology used to generate
them as well as inherent variation within samples. The presence of high levels
of noise can hamper discovery of patterns and dynamics encapsulated by these
networks. Here we propose Network Enhancement (NE), a novel method for
improving the signal-to-noise ratio of undirected, weighted networks, and
thereby improving the performance of downstream analysis. NE applies a novel
operator that induces sparsity and leverages higher-order network structures to
remove weak edges and enhance real connections. This iterative approach has a
closed-form solution at convergence with desirable performance properties. We
demonstrate the effectiveness of NE in denoising biological networks for
several challenging yet important problems. Our experiments show that NE
improves gene function prediction by denoising interaction networks from 22
human tissues. Further, we use NE to interpret noisy Hi-C contact maps from the
human genome and demonstrate its utility across varying degrees of data
quality. Finally, when applied to fine-grained species identification, NE
outperforms alternative approaches by a significant margin. Taken together, our
results indicate that NE is widely applicable for denoising weighted biological
networks, especially when they contain high levels of noise.
-
Logical models offer a simple but powerful means to understand the complex
dynamics of biochemical regulation, without the need to estimate kinetic
parameters. However, even simple automata components can lead to collective
dynamics that are computationally intractable when aggregated into networks. In
previous work we demonstrated that automata network models of biochemical
regulation are highly canalizing, whereby many variable states and their
groupings are redundant (Marques-Pita and Rocha, 2013). The precise charting
and measurement of such canalization simplifies these models, making even very
large networks amenable to analysis. Moreover, canalization plays an important
role in the control, robustness, modularity and criticality of Boolean network
dynamics, especially those used to model biochemical regulation (Gates and
Rocha, 2016; Gates et al., 2016; Manicka, 2017). Here we describe a new
publicly-available Python package that provides the necessary tools to extract,
measure, and visualize canalizing redundancy present in Boolean network models.
It extracts the pathways most effective in controlling dynamics in these
models, including their effective graph and dynamics canalizing map, as well as
other tools to uncover minimum sets of control variables.
-
Infection by many viruses begins with fusion of viral and cellular lipid
membranes, followed by entry of viral contents into the target cell and
ultimately, after many biochemical steps, integration of viral DNA into that of
the host cell. The early steps of membrane fusion and viral capsid entry are
mediated by adsorption to the cell surface, and receptor and coreceptor
binding. HIV-1 specifically targets CD4+ helper T-cells of the human immune
system and binds to the receptor CD4 and coreceptor CCR5 before fusion is
initiated. Previous experiments have been performed using a cell line
(293-Affinofile) in which the expression of CD4 and CCR5 concentration were
independently controlled. After exposure to HIV-1 of various strains, the
resulting infectivity was measured through the fraction of infected cells. To
design and evaluate the effectiveness of drug therapies that target the
inhibition of the entry processes, an accurate functional relationship between
the CD4/CCR5 concentrations and infectivity is desired in order to more
quantitatively analyze experimental data. We propose three kinetic models
describing the possible mechanistic processes involved in HIV entry and fit
their predictions to infectivity measurements, contrasting and comparing
different outcomes. Our approach allows interpretation of the clustering of
infectivity of different strains of HIV-1 in the space of mechanistic kinetic
parameters. Our model fitting also allows inference of nontrivial
stoichiometries of receptor and coreceptor binding and provides a framework
through which to quantitatively investigate the effectiveness of fusion
inhibitors and neutralizing antibodies.





 Due to the scarcity of quantitative details about biological phenomena, quantitative modeling in systems biology can be compromised, especially at the subcellular scale. One way to get around this is qualitative modeling because it requires few to no quantitative information. One of the most popular qualitative modeling approaches is the Boolean network formalism. However, Boolean models allow variables to take only two values, which can be too simplistic in some cases. The present work proposes a modeling approach derived from Boolean networks where continuous logical operators are used and where edges can be tuned. Using continuous logical operators allows variables to be more finely valued while remaining qualitative. To consider that some biological interactions can be slower or weaker than other ones, edge states are also computed in order to modulate in speed and strength the signal they convey. The proposed formalism is illustrated on a toy network coming from the epidermal growth factor receptor signaling pathway. The obtained simulations show that continuous results are produced, thus allowing finer analysis. The simulations also show that modulating the signal conveyed by the edges allows to incorporate knowledge about the interactions they model. The goal is to provide enhancements in the ability of qualitative models to simulate the dynamics of biological networks while limiting the need of quantitative information.
Due to the scarcity of quantitative details about biological phenomena, quantitative modeling in systems biology can be compromised, especially at the subcellular scale. One way to get around this is qualitative modeling because it requires few to no quantitative information. One of the most popular qualitative modeling approaches is the Boolean network formalism. However, Boolean models allow variables to take only two values, which can be too simplistic in some cases. The present work proposes a modeling approach derived from Boolean networks where continuous logical operators are used and where edges can be tuned. Using continuous logical operators allows variables to be more finely valued while remaining qualitative. To consider that some biological interactions can be slower or weaker than other ones, edge states are also computed in order to modulate in speed and strength the signal they convey. The proposed formalism is illustrated on a toy network coming from the epidermal growth factor receptor signaling pathway. The obtained simulations show that continuous results are produced, thus allowing finer analysis. The simulations also show that modulating the signal conveyed by the edges allows to incorporate knowledge about the interactions they model. The goal is to provide enhancements in the ability of qualitative models to simulate the dynamics of biological networks while limiting the need of quantitative information.
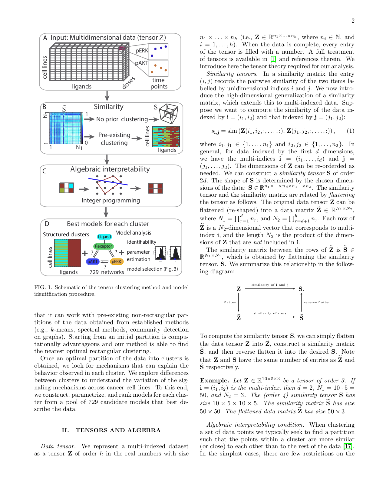



 We introduce a tensor-based clustering method to extract sparse, low-dimensional structure from high-dimensional, multi-indexed datasets. This framework is designed to enable detection of clusters of data in the presence of structural requirements which we encode as algebraic constraints in a linear program. Our clustering method is general and can be tailored to a variety of applications in science and industry. We illustrate our method on a collection of experiments measuring the response of genetically diverse breast cancer cell lines to an array of ligands. Each experiment consists of a cell line-ligand combination, and contains time-course measurements of the early-signalling kinases MAPK and AKT at two different ligand dose levels. By imposing appropriate structural constraints and respecting the multi-indexed structure of the data, the analysis of clusters can be optimized for biological interpretation and therapeutic understanding. We then perform a systematic, large-scale exploration of mechanistic models of MAPK-AKT crosstalk for each cluster. This analysis allows us to quantify the heterogeneity of breast cancer cell subtypes, and leads to hypotheses about the signalling mechanisms that mediate the response of the cell lines to ligands.
We introduce a tensor-based clustering method to extract sparse, low-dimensional structure from high-dimensional, multi-indexed datasets. This framework is designed to enable detection of clusters of data in the presence of structural requirements which we encode as algebraic constraints in a linear program. Our clustering method is general and can be tailored to a variety of applications in science and industry. We illustrate our method on a collection of experiments measuring the response of genetically diverse breast cancer cell lines to an array of ligands. Each experiment consists of a cell line-ligand combination, and contains time-course measurements of the early-signalling kinases MAPK and AKT at two different ligand dose levels. By imposing appropriate structural constraints and respecting the multi-indexed structure of the data, the analysis of clusters can be optimized for biological interpretation and therapeutic understanding. We then perform a systematic, large-scale exploration of mechanistic models of MAPK-AKT crosstalk for each cluster. This analysis allows us to quantify the heterogeneity of breast cancer cell subtypes, and leads to hypotheses about the signalling mechanisms that mediate the response of the cell lines to ligands.


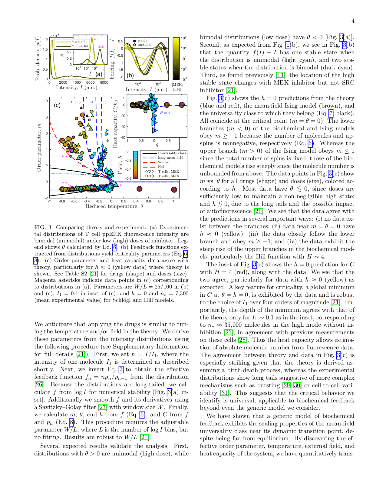

 We map a class of well-mixed stochastic models of biochemical feedback in steady state to the mean-field Ising model near the critical point. The mapping provides an effective temperature, magnetic field, order parameter, and heat capacity that can be extracted from biological data without fitting or knowledge of the underlying molecular details. We demonstrate this procedure on fluorescence data from mouse T cells, which reveals distinctions between how the cells respond to different drugs. We also show that the heat capacity allows inference of absolute molecule number from fluorescence intensity. We explain this result in terms of the underlying fluctuations and demonstrate the generality of our work.
We map a class of well-mixed stochastic models of biochemical feedback in steady state to the mean-field Ising model near the critical point. The mapping provides an effective temperature, magnetic field, order parameter, and heat capacity that can be extracted from biological data without fitting or knowledge of the underlying molecular details. We demonstrate this procedure on fluorescence data from mouse T cells, which reveals distinctions between how the cells respond to different drugs. We also show that the heat capacity allows inference of absolute molecule number from fluorescence intensity. We explain this result in terms of the underlying fluctuations and demonstrate the generality of our work.




 The advent of high--throughput transcription profiling technologies has enabled identification of genes and pathways associated with disease, providing new avenues for precision medicine. A key challenge is to analyze this data in the context of the regulatory networks and pathways that control cellular processes, while still obtaining insights that can be used to design new diagnostic and therapeutic interventions. While classical differential expression analysis provides specific and hence targetable gene-level insights, it does not include any systems-level information. On the other hand, pathway analyses integrate systems-level information with expression data, but are often limited in their ability to indicate specific molecular targets. We introduce GeneSurrounder, an analysis method that takes into account the complex structure of interaction networks to identify specific genes that disrupt pathway activity in a disease-specific manner. GeneSurrounder integrates transcriptomic data and pathway network information in a novel two-step procedure to detect genes that (i) appear to influence the expression of other genes local to it in the network and (ii) are part of a subnetwork of differentially expressed genes. Combined, this evidence can be used to pinpoint specific genes that have a mechanistic role in the phenotype of interest. Applying GeneSurrounder to three distinct ovarian cancer studies using a global KEGG network, we show that our method is able to identify biologically relevant genes and genes missed by single-gene association tests, integrate pathway and expression data, and yield more consistent results across multiple studies of the same phenotype than competing methods.
The advent of high--throughput transcription profiling technologies has enabled identification of genes and pathways associated with disease, providing new avenues for precision medicine. A key challenge is to analyze this data in the context of the regulatory networks and pathways that control cellular processes, while still obtaining insights that can be used to design new diagnostic and therapeutic interventions. While classical differential expression analysis provides specific and hence targetable gene-level insights, it does not include any systems-level information. On the other hand, pathway analyses integrate systems-level information with expression data, but are often limited in their ability to indicate specific molecular targets. We introduce GeneSurrounder, an analysis method that takes into account the complex structure of interaction networks to identify specific genes that disrupt pathway activity in a disease-specific manner. GeneSurrounder integrates transcriptomic data and pathway network information in a novel two-step procedure to detect genes that (i) appear to influence the expression of other genes local to it in the network and (ii) are part of a subnetwork of differentially expressed genes. Combined, this evidence can be used to pinpoint specific genes that have a mechanistic role in the phenotype of interest. Applying GeneSurrounder to three distinct ovarian cancer studies using a global KEGG network, we show that our method is able to identify biologically relevant genes and genes missed by single-gene association tests, integrate pathway and expression data, and yield more consistent results across multiple studies of the same phenotype than competing methods.




 This paper proposes a control theoretic framework to model and analyze the self-organized pattern formation of molecular concentrations in biomolecular communication networks, emerging applications in synthetic biology. In biomolecular communication networks, bionanomachines, or biological cells, communicate with each other using a cell-to-cell communication mechanism mediated by a diffusible signaling molecule, thereby the dynamics of molecular concentrations are approximately modeled as a reaction-diffusion system with a single diffuser. We first introduce a feedback model representation of the reaction-diffusion system and provide a systematic local stability/instability analysis tool using the root locus of the feedback system. The instability analysis then allows us to analytically derive the conditions for the self-organized spatial pattern formation, or Turing pattern formation, of the bionanomachines. We propose a novel synthetic biocircuit motif called activator-repressor-diffuser system and show that it is one of the minimum biomolecular circuits that admit self-organized patterns over cell population.
This paper proposes a control theoretic framework to model and analyze the self-organized pattern formation of molecular concentrations in biomolecular communication networks, emerging applications in synthetic biology. In biomolecular communication networks, bionanomachines, or biological cells, communicate with each other using a cell-to-cell communication mechanism mediated by a diffusible signaling molecule, thereby the dynamics of molecular concentrations are approximately modeled as a reaction-diffusion system with a single diffuser. We first introduce a feedback model representation of the reaction-diffusion system and provide a systematic local stability/instability analysis tool using the root locus of the feedback system. The instability analysis then allows us to analytically derive the conditions for the self-organized spatial pattern formation, or Turing pattern formation, of the bionanomachines. We propose a novel synthetic biocircuit motif called activator-repressor-diffuser system and show that it is one of the minimum biomolecular circuits that admit self-organized patterns over cell population.




 In this paper we investigate the complexity of model selection and model testing for dynamical systems with toric steady states. Such systems frequently arise in the study of chemical reaction networks. We do this by formulating these tasks as a constrained optimization problem in Euclidean space. This optimization problem is known as a Euclidean distance problem; the complexity of solving this problem is measured by an invariant called the Euclidean distance (ED) degree. We determine closed-form expressions for the ED degree of the steady states of several families of chemical reaction networks with toric steady states and arbitrarily many reactions. To illustrate the utility of this work we show how the ED degree can be used as a tool for estimating the computational cost of solving the model testing and model selection problems.
In this paper we investigate the complexity of model selection and model testing for dynamical systems with toric steady states. Such systems frequently arise in the study of chemical reaction networks. We do this by formulating these tasks as a constrained optimization problem in Euclidean space. This optimization problem is known as a Euclidean distance problem; the complexity of solving this problem is measured by an invariant called the Euclidean distance (ED) degree. We determine closed-form expressions for the ED degree of the steady states of several families of chemical reaction networks with toric steady states and arbitrarily many reactions. To illustrate the utility of this work we show how the ED degree can be used as a tool for estimating the computational cost of solving the model testing and model selection problems.


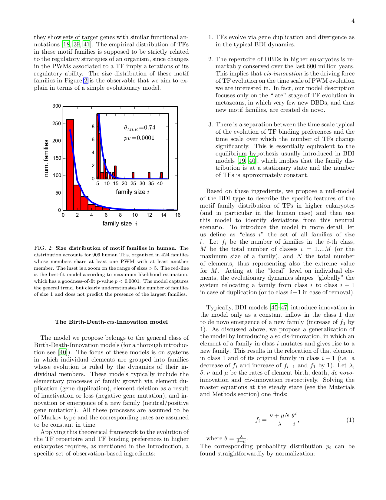

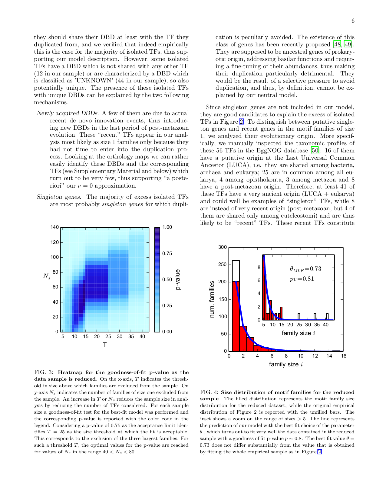 Transcription factors (TFs) exert their regulatory action by binding to DNA with specific sequence preferences. However, different TFs can partially share their binding sequences due to their common evolutionary origin. This `redundancy' of binding defines a way of organizing TFs in `motif families' by grouping TFs with similar binding preferences. Since these ultimately define the TF target genes, the motif family organization entails information about the structure of transcriptional regulation as it has been shaped by evolution. Focusing on the human TF repertoire, we show that a one-parameter evolutionary model of the Birth-Death-Innovation type can explain the TF empirical ripartition in motif families, and allows to highlight the relevant evolutionary forces at the origin of this organization. Moreover, the model allows to pinpoint few deviations from the neutral scenario it assumes: three over-expanded families (including HOX and FOX genes), a set of `singleton' TFs for which duplication seems to be selected against, and a higher-than-average rate of diversification of the binding preferences of TFs with a Zinc Finger DNA binding domain. Finally, a comparison of the TF motif family organization in different eukaryotic species suggests an increase of redundancy of binding with organism complexity.
Transcription factors (TFs) exert their regulatory action by binding to DNA with specific sequence preferences. However, different TFs can partially share their binding sequences due to their common evolutionary origin. This `redundancy' of binding defines a way of organizing TFs in `motif families' by grouping TFs with similar binding preferences. Since these ultimately define the TF target genes, the motif family organization entails information about the structure of transcriptional regulation as it has been shaped by evolution. Focusing on the human TF repertoire, we show that a one-parameter evolutionary model of the Birth-Death-Innovation type can explain the TF empirical ripartition in motif families, and allows to highlight the relevant evolutionary forces at the origin of this organization. Moreover, the model allows to pinpoint few deviations from the neutral scenario it assumes: three over-expanded families (including HOX and FOX genes), a set of `singleton' TFs for which duplication seems to be selected against, and a higher-than-average rate of diversification of the binding preferences of TFs with a Zinc Finger DNA binding domain. Finally, a comparison of the TF motif family organization in different eukaryotic species suggests an increase of redundancy of binding with organism complexity.




 Controlling stochastic reactions networks is a challenging problem with important implications in various fields such as systems and synthetic biology. Various regulation motifs have been discovered or posited over the recent years, the most recent one being the so-called Antithetic Integral Control (AIC) motif in Briat et al. (Cell Systems, 2016). Several favorable properties for the AIC motif have been demonstrated for classes of reaction networks that satisfy certain irreducibility, ergodicity and output controllability conditions. Here we address the problem of verifying these conditions for large sets of reaction networks with fixed topology using two different approaches. The first one is quantitative and relies on the notion of interval matrices while the second one is qualitative and is based on sign properties of matrices. The obtained results lie in the same spirit as those obtained in Briat et al. (Cell Systems, 2016) where properties of reaction networks are independently characterized in terms of control theoretic concepts, linear programming conditions and graph theoretic conditions.
Controlling stochastic reactions networks is a challenging problem with important implications in various fields such as systems and synthetic biology. Various regulation motifs have been discovered or posited over the recent years, the most recent one being the so-called Antithetic Integral Control (AIC) motif in Briat et al. (Cell Systems, 2016). Several favorable properties for the AIC motif have been demonstrated for classes of reaction networks that satisfy certain irreducibility, ergodicity and output controllability conditions. Here we address the problem of verifying these conditions for large sets of reaction networks with fixed topology using two different approaches. The first one is quantitative and relies on the notion of interval matrices while the second one is qualitative and is based on sign properties of matrices. The obtained results lie in the same spirit as those obtained in Briat et al. (Cell Systems, 2016) where properties of reaction networks are independently characterized in terms of control theoretic concepts, linear programming conditions and graph theoretic conditions.

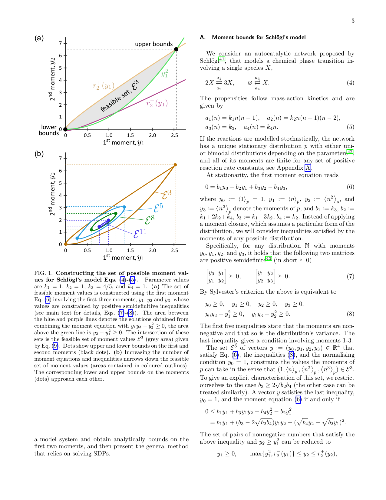
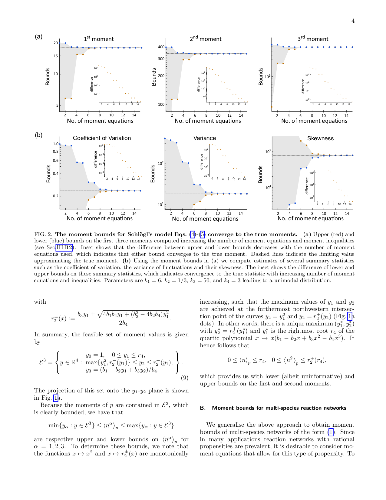

 The stochastic dynamics of biochemical networks are usually modelled with the chemical master equation (CME). The stationary distributions of CMEs are seldom solvable analytically, and numerical methods typically produce estimates with uncontrolled errors. Here, we introduce mathematical programming approaches that yield approximations of these distributions with computable error bounds which enable the verification of their accuracy. First, we use semidefinite programming to compute increasingly tighter upper and lower bounds on the moments of the stationary distributions for networks with rational propensities. Second, we use these moment bounds to formulate linear programs that yield convergent upper and lower bounds on the stationary distributions themselves, their marginals and stationary averages. The bounds obtained also provide a computational test for the uniqueness of the distribution. In the unique case, the bounds form an approximation of the stationary distribution with a computable bound on its error. In the non-unique case, our approach yields converging approximations of the ergodic distributions. We illustrate our methodology through several biochemical examples taken from the literature: Schl\"ogl's model for a chemical bifurcation, a two-dimensional toggle switch, a model for bursty gene expression, and a dimerisation model with multiple stationary distributions.
The stochastic dynamics of biochemical networks are usually modelled with the chemical master equation (CME). The stationary distributions of CMEs are seldom solvable analytically, and numerical methods typically produce estimates with uncontrolled errors. Here, we introduce mathematical programming approaches that yield approximations of these distributions with computable error bounds which enable the verification of their accuracy. First, we use semidefinite programming to compute increasingly tighter upper and lower bounds on the moments of the stationary distributions for networks with rational propensities. Second, we use these moment bounds to formulate linear programs that yield convergent upper and lower bounds on the stationary distributions themselves, their marginals and stationary averages. The bounds obtained also provide a computational test for the uniqueness of the distribution. In the unique case, the bounds form an approximation of the stationary distribution with a computable bound on its error. In the non-unique case, our approach yields converging approximations of the ergodic distributions. We illustrate our methodology through several biochemical examples taken from the literature: Schl\"ogl's model for a chemical bifurcation, a two-dimensional toggle switch, a model for bursty gene expression, and a dimerisation model with multiple stationary distributions.


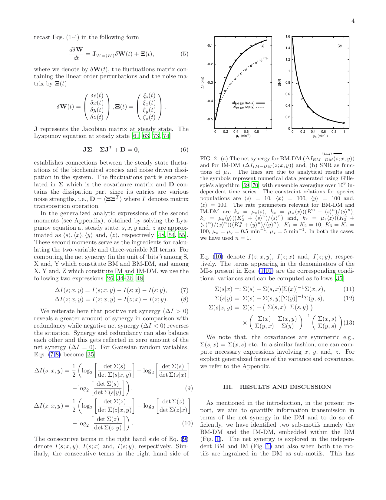

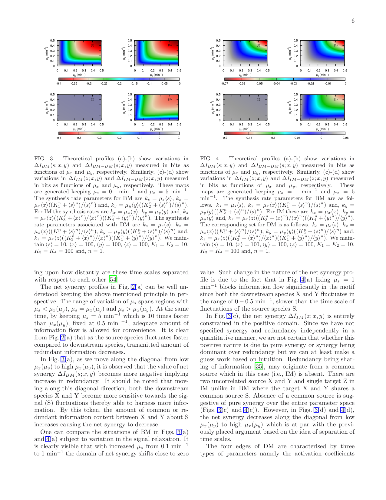 The formalism of partial information decomposition provides independent or non-overlapping components constituting total information content provided by a set of source variables about the target variable. These components are recognised as unique information, synergistic information and, redundant information. The metric of net synergy, conceived as the difference between synergistic and redundant information, is capable of detecting synergy, redundancy and, information independence among stochastic variables. And it can be quantified, as it is done here, using appropriate combinations of different Shannon mutual information terms. Utilisation of such a metric in network motifs with the nodes representing different biochemical species, involved in information sharing, uncovers rich store for interesting results. In the current study, we make use of this formalism to obtain a comprehensive understanding of the relative information processing mechanism in a diamond motif and two of its sub-motifs namely bifurcation and integration motif embedded within the diamond motif. The emerging patterns of synergy and redundancy and their effective contribution towards ensuring high fidelity information transmission are duly compared in the sub-motifs and independent motifs (bifurcation and integration). In this context, the crucial roles played by various time scales and activation coefficients in the network topologies are especially emphasised. We show that the origin of synergy and redundancy in information transmission can be physically justified by decomposing diamond motif into bifurcation and integration motif.
The formalism of partial information decomposition provides independent or non-overlapping components constituting total information content provided by a set of source variables about the target variable. These components are recognised as unique information, synergistic information and, redundant information. The metric of net synergy, conceived as the difference between synergistic and redundant information, is capable of detecting synergy, redundancy and, information independence among stochastic variables. And it can be quantified, as it is done here, using appropriate combinations of different Shannon mutual information terms. Utilisation of such a metric in network motifs with the nodes representing different biochemical species, involved in information sharing, uncovers rich store for interesting results. In the current study, we make use of this formalism to obtain a comprehensive understanding of the relative information processing mechanism in a diamond motif and two of its sub-motifs namely bifurcation and integration motif embedded within the diamond motif. The emerging patterns of synergy and redundancy and their effective contribution towards ensuring high fidelity information transmission are duly compared in the sub-motifs and independent motifs (bifurcation and integration). In this context, the crucial roles played by various time scales and activation coefficients in the network topologies are especially emphasised. We show that the origin of synergy and redundancy in information transmission can be physically justified by decomposing diamond motif into bifurcation and integration motif.




 Sensitivity analysis of biochemical reactions aims at quantifying the dependence of the reaction dynamics on the reaction rates. The computation of the parameter sensitivities, however, poses many computational challenges when taking stochastic noise into account. This paper proposes a new finite difference method for efficiently computing sensitivities of biochemical reactions. We employ propensity bounds of reactions to couple the simulation of the nominal and perturbed processes. The exactness of the simulation is reserved by applying the rejection-based mechanism. For each simulation step, the nominal and perturbed processes under our coupling strategy are synchronized and often jump together, increasing their positive correlation and hence reducing the variance of the estimator. The distinctive feature of our approach in comparison with existing coupling approaches is that it only needs to maintain a single data structure storing propensity bounds of reactions during the simulation of the nominal and perturbed processes. Our approach allows to computing sensitivities of many reaction rates simultaneously. Moreover, the data structure does not require to be updated frequently, hence improving the computational cost. This feature is especially useful when applied to large reaction networks. We benchmark our method on biological reaction models to prove its applicability and efficiency.
Sensitivity analysis of biochemical reactions aims at quantifying the dependence of the reaction dynamics on the reaction rates. The computation of the parameter sensitivities, however, poses many computational challenges when taking stochastic noise into account. This paper proposes a new finite difference method for efficiently computing sensitivities of biochemical reactions. We employ propensity bounds of reactions to couple the simulation of the nominal and perturbed processes. The exactness of the simulation is reserved by applying the rejection-based mechanism. For each simulation step, the nominal and perturbed processes under our coupling strategy are synchronized and often jump together, increasing their positive correlation and hence reducing the variance of the estimator. The distinctive feature of our approach in comparison with existing coupling approaches is that it only needs to maintain a single data structure storing propensity bounds of reactions during the simulation of the nominal and perturbed processes. Our approach allows to computing sensitivities of many reaction rates simultaneously. Moreover, the data structure does not require to be updated frequently, hence improving the computational cost. This feature is especially useful when applied to large reaction networks. We benchmark our method on biological reaction models to prove its applicability and efficiency.


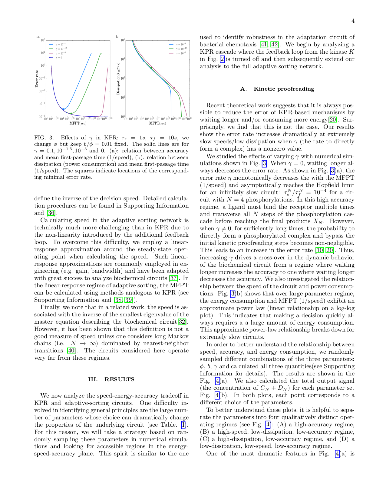
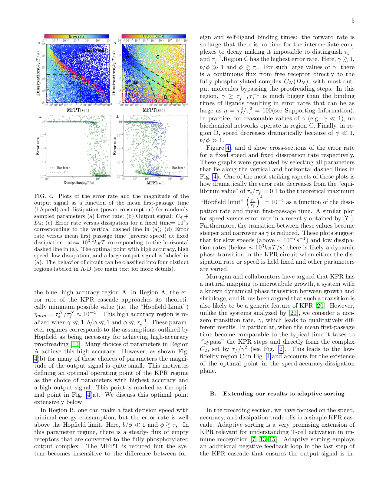
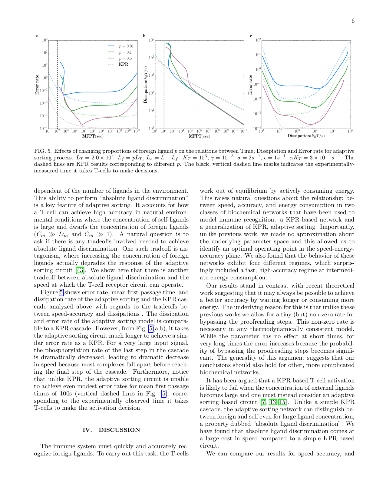 In the immune system, T cells can quickly discriminate between foreign and self ligands with high accuracy. There is evidence that T-cells achieve this remarkable performance utilizing a network architecture based on a generalization of kinetic proofreading (KPR). KPR-based mechanisms actively consume energy to increase the specificity beyond what is possible in equilibrium.An important theoretical question that arises is to understand the trade-offs and fundamental limits on accuracy, speed, and dissipation (energy consumption) in KPR and its generalization. Here, we revisit this question through numerical simulations where we simultaneously measure the speed, accuracy, and energy consumption of the KPR and adaptive sorting networks for different parameter choices. Our simulations highlight the existence of a 'feasible operating regime' in the speed-energy-accuracy plane where T-cells can quickly differentiate between foreign and self ligands at reasonable energy expenditure. We give general arguments for why we expect this feasible operating regime to be a generic property of all KPR-based biochemical networks and discuss implications for our understanding of the T cell receptor circuit.
In the immune system, T cells can quickly discriminate between foreign and self ligands with high accuracy. There is evidence that T-cells achieve this remarkable performance utilizing a network architecture based on a generalization of kinetic proofreading (KPR). KPR-based mechanisms actively consume energy to increase the specificity beyond what is possible in equilibrium.An important theoretical question that arises is to understand the trade-offs and fundamental limits on accuracy, speed, and dissipation (energy consumption) in KPR and its generalization. Here, we revisit this question through numerical simulations where we simultaneously measure the speed, accuracy, and energy consumption of the KPR and adaptive sorting networks for different parameter choices. Our simulations highlight the existence of a 'feasible operating regime' in the speed-energy-accuracy plane where T-cells can quickly differentiate between foreign and self ligands at reasonable energy expenditure. We give general arguments for why we expect this feasible operating regime to be a generic property of all KPR-based biochemical networks and discuss implications for our understanding of the T cell receptor circuit.


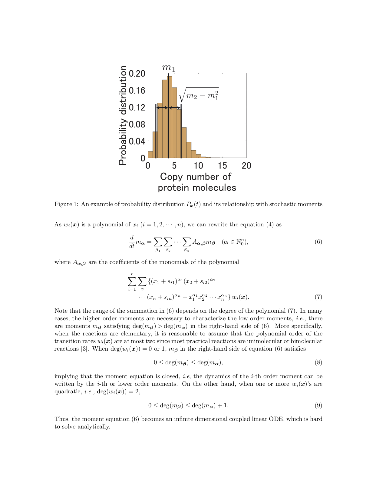

 Model-based prediction of stochastic noise in biomolecular reactions often resorts to approximation with unknown precision. As a result, unexpected stochastic fluctuation causes a headache for the designers of biomolecular circuits. This paper proposes a convex optimization approach to quantifying the steady state moments of molecular copy counts with theoretical rigor. We show that the stochastic moments lie in a convex semi-algebraic set specified by linear matrix inequalities. Thus, the upper and the lower bounds of some moments can be computed by a semidefinite program. Using a protein dimerization process as an example, we demonstrate that the proposed method can precisely predict the mean and the variance of the copy number of the monomer protein.
Model-based prediction of stochastic noise in biomolecular reactions often resorts to approximation with unknown precision. As a result, unexpected stochastic fluctuation causes a headache for the designers of biomolecular circuits. This paper proposes a convex optimization approach to quantifying the steady state moments of molecular copy counts with theoretical rigor. We show that the stochastic moments lie in a convex semi-algebraic set specified by linear matrix inequalities. Thus, the upper and the lower bounds of some moments can be computed by a semidefinite program. Using a protein dimerization process as an example, we demonstrate that the proposed method can precisely predict the mean and the variance of the copy number of the monomer protein.




 Investigations of topological uniqueness of gene interaction networks in cancer cells are essential for understanding this disease. Based on the random matrix theory, we study the distribution of the nearest neighbor level spacings $P(s)$ of interaction matrices for gene networks in human cancer cells. The interaction matrices are computed using the Cancer Network Galaxy (TCNG) database, which is a repository of gene interactions inferred by a Bayesian network model. 256 NCBI GEO entries regarding gene expressions in human cancer cells have been selected for the Bayesian network calculations in TCNG. We observe the Wigner distribution of $P(s)$ when the gene networks are dense networks that have more than $\sim 38,000$ edges. In the opposite case, when the networks have smaller numbers of edges, the distribution $P(s)$ becomes the Poisson distribution. We investigate relevance of $P(s)$ both to the size of the networks and to edge frequencies that manifest reliance of the inferred gene interactions.
Investigations of topological uniqueness of gene interaction networks in cancer cells are essential for understanding this disease. Based on the random matrix theory, we study the distribution of the nearest neighbor level spacings $P(s)$ of interaction matrices for gene networks in human cancer cells. The interaction matrices are computed using the Cancer Network Galaxy (TCNG) database, which is a repository of gene interactions inferred by a Bayesian network model. 256 NCBI GEO entries regarding gene expressions in human cancer cells have been selected for the Bayesian network calculations in TCNG. We observe the Wigner distribution of $P(s)$ when the gene networks are dense networks that have more than $\sim 38,000$ edges. In the opposite case, when the networks have smaller numbers of edges, the distribution $P(s)$ becomes the Poisson distribution. We investigate relevance of $P(s)$ both to the size of the networks and to edge frequencies that manifest reliance of the inferred gene interactions.

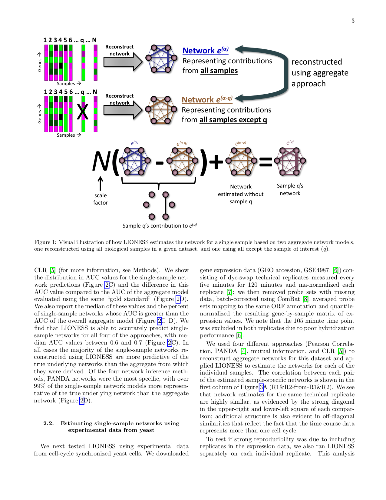
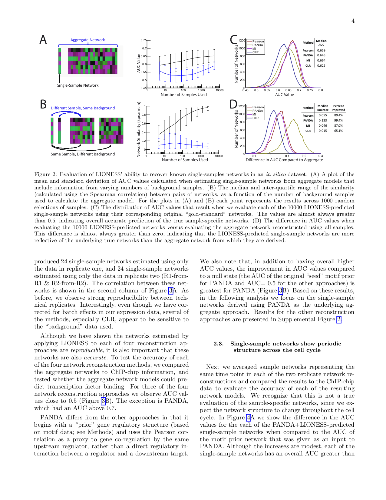

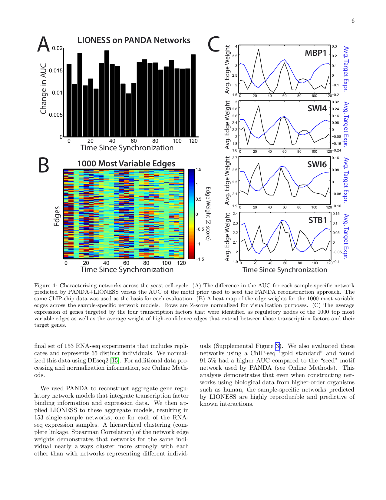 Biological systems are driven by intricate interactions among the complex array of molecules that comprise the cell. Many methods have been developed to reconstruct network models of those interactions. These methods often draw on large numbers of samples with measured gene expression profiles to infer connections between genes (or gene products). The result is an aggregate network model representing a single estimate for the likelihood of each interaction, or "edge," in the network. While informative, aggregate models fail to capture the heterogeneity that is represented in any population. Here we propose a method to reverse engineer sample-specific networks from aggregate network models. We demonstrate the accuracy and applicability of our approach in several data sets, including simulated data, microarray expression data from synchronized yeast cells, and RNA-seq data collected from human lymphoblastoid cell lines. We show that these sample-specific networks can be used to study changes in network topology across time and to characterize shifts in gene regulation that may not be apparent in expression data. We believe the ability to generate sample-specific networks will greatly facilitate the application of network methods to the increasingly large, complex, and heterogeneous multi-omic data sets that are currently being generated, and ultimately support the emerging field of precision network medicine.
Biological systems are driven by intricate interactions among the complex array of molecules that comprise the cell. Many methods have been developed to reconstruct network models of those interactions. These methods often draw on large numbers of samples with measured gene expression profiles to infer connections between genes (or gene products). The result is an aggregate network model representing a single estimate for the likelihood of each interaction, or "edge," in the network. While informative, aggregate models fail to capture the heterogeneity that is represented in any population. Here we propose a method to reverse engineer sample-specific networks from aggregate network models. We demonstrate the accuracy and applicability of our approach in several data sets, including simulated data, microarray expression data from synchronized yeast cells, and RNA-seq data collected from human lymphoblastoid cell lines. We show that these sample-specific networks can be used to study changes in network topology across time and to characterize shifts in gene regulation that may not be apparent in expression data. We believe the ability to generate sample-specific networks will greatly facilitate the application of network methods to the increasingly large, complex, and heterogeneous multi-omic data sets that are currently being generated, and ultimately support the emerging field of precision network medicine.




 Phenotypical variability in the absence of genetic variation often reflects complex energetic landscapes associated with underlying gene regulatory networks (GRNs). In this view, different phenotypes are associated with alternative states of complex nonlinear systems: stable attractors in deterministic models or modes of stationary distributions in stochastic descriptions. We provide theoretical and practical characterizations of these landscapes, specifically focusing on stochastic slow promoter kinetics, a time scale relevant when transcription factor binding and unbinding are affected by epigenetic processes like DNA methylation and chromatin remodeling. In this case, largely unexplored except for numerical simulations, adiabatic approximations of promoter kinetics are not appropriate. In contrast to the existing literature, we provide rigorous analytic characterizations of multiple modes. A general formal approach gives insight into the influence of parameters and the prediction of how changes in GRN wiring, for example through mutations or artificial interventions, impact the possible number, location, and likelihood of alternative states. We adapt tools from the mathematical field of singular perturbation theory to represent stationary distributions of Chemical Master Equations for GRNs as mixtures of Poisson distributions and obtain explicit formulas for the locations and probabilities of metastable states as a function of the parameters describing the system. As illustrations, the theory is used to tease out the role of cooperative binding in stochastic models in comparison to deterministic models, and applications are given to various model systems, such as toggle switches in isolation or in communicating populations, a synthetic oscillator and a trans-differentiation network.
Phenotypical variability in the absence of genetic variation often reflects complex energetic landscapes associated with underlying gene regulatory networks (GRNs). In this view, different phenotypes are associated with alternative states of complex nonlinear systems: stable attractors in deterministic models or modes of stationary distributions in stochastic descriptions. We provide theoretical and practical characterizations of these landscapes, specifically focusing on stochastic slow promoter kinetics, a time scale relevant when transcription factor binding and unbinding are affected by epigenetic processes like DNA methylation and chromatin remodeling. In this case, largely unexplored except for numerical simulations, adiabatic approximations of promoter kinetics are not appropriate. In contrast to the existing literature, we provide rigorous analytic characterizations of multiple modes. A general formal approach gives insight into the influence of parameters and the prediction of how changes in GRN wiring, for example through mutations or artificial interventions, impact the possible number, location, and likelihood of alternative states. We adapt tools from the mathematical field of singular perturbation theory to represent stationary distributions of Chemical Master Equations for GRNs as mixtures of Poisson distributions and obtain explicit formulas for the locations and probabilities of metastable states as a function of the parameters describing the system. As illustrations, the theory is used to tease out the role of cooperative binding in stochastic models in comparison to deterministic models, and applications are given to various model systems, such as toggle switches in isolation or in communicating populations, a synthetic oscillator and a trans-differentiation network.




 We consider stochastically modeled reaction networks and prove that if a constant solution to the Kolmogorov forward equation decays fast enough relatively to the transition rates, then the model is non-explosive. In particular, complex balanced reaction networks are non-explosive.
We consider stochastically modeled reaction networks and prove that if a constant solution to the Kolmogorov forward equation decays fast enough relatively to the transition rates, then the model is non-explosive. In particular, complex balanced reaction networks are non-explosive.