-
We propose a general framework for nonasymptotic covariance matrix estimation
making use of concentration inequality-based confidence sets. We specify this
framework for the estimation of large sparse covariance matrices through
incorporation of past thresholding estimators with key emphasis on support
recovery. This technique goes beyond past results for thresholding estimators
by allowing for a wide range of distributional assumptions beyond merely
sub-Gaussian tails. This methodology can furthermore be adapted to a wide range
of other estimators and settings. The usage of nonasymptotic dimension-free
confidence sets yields good theoretical performance. Through extensive
simulations, it is demonstrated to have superior performance when compared with
other such methods. In the context of support recovery, we are able to specify
a false positive rate and optimize to maximize the true recoveries.
-
Calibration of hydrological time-series models is a challenging task since
these models give a wide spectrum of output series and calibration procedures
require significant amount of time. From a statistical standpoint, this model
parameter estimation problem simplifies to finding an inverse solution of a
computer model that generates pre-specified time-series output (i.e., realistic
output series). In this paper, we propose a modified history matching approach
for calibrating the time-series rainfall-runoff models with respect to the real
data collected from the state of Georgia, USA. We present the methodology and
illustrate the application of the algorithm by carrying a simulation study and
the two case studies. Several goodness-of-fit statistics were calculated to
assess the model performance. The results showed that the proposed history
matching algorithm led to a significant improvement, of 30% and 14% (in terms
of root mean squared error) and 26% and 118% (in terms of peak percent
threshold statistics), for the two case-studies with Matlab-Simulink and SWAT
models, respectively.
-
In this article, we present data-subsetting algorithms that allow for the
approximate and scalable implementation of the Bayesian bootstrap. They are
analogous to two existing algorithms in the frequentist literature: the bag of
little bootstraps (Kleiner et al., 2014) and the subsampled double bootstrap
(SDB; Sengupta et al., 2016). Our algorithms have appealing theoretical and
computational properties that are comparable to those of their frequentist
counterparts. Additionally, we provide a strategy for performing lossless
inference for a class of functionals of the Bayesian bootstrap, and briefly
introduce extensions to the Dirichlet Process.
-
We investigate a new sampling scheme aimed at improving the performance of
particle filters whenever (a) there is a significant mismatch between the
assumed model dynamics and the actual system, or (b) the posterior probability
tends to concentrate in relatively small regions of the state space. The
proposed scheme pushes some particles towards specific regions where the
likelihood is expected to be high, an operation known as nudging in the
geophysics literature. We re-interpret nudging in a form applicable to any
particle filtering scheme, as it does not involve any changes in the rest of
the algorithm. Since the particles are modified, but the importance weights do
not account for this modification, the use of nudging leads to additional bias
in the resulting estimators. However, we prove analytically that nudged
particle filters can still attain asymptotic convergence with the same error
rates as conventional particle methods. Simple analysis also yields an
alternative interpretation of the nudging operation that explains its
robustness to model errors. Finally, we show numerical results that illustrate
the improvements that can be attained using the proposed scheme. In particular,
we present nonlinear tracking examples with synthetic data and a model
inference example using real-world financial data.
-
Statistical analyses of directional or angular data have applications in a
variety of fields, such as geology, meteorology and bioinformatics. There is
substantial literature on descriptive and inferential techniques for univariate
angular data, with the bivariate (or more generally, multivariate) cases
receiving more attention in recent years. More specifically, the bivariate
wrapped normal, von Mises sine and von Mises cosine distributions, and mixtures
thereof, have been proposed for practical use. However, there is a lack of
software implementing these distributions and the associated inferential
techniques. In this article, we introduce BAMBI, an R package for analyzing
bivariate (and univariate) angular data. We implement random data generation,
density evaluation, and computation of theoretical summary measures (variances
and correlation coefficients) for the three aforementioned bivariate angular
distributions, as well as two univariate angular distributions: the univariate
wrapped normal and the univariate von Mises distribution. The major
contribution of BAMBI to statistical computing is in providing Bayesian methods
for modeling angular data using finite mixtures of these distributions. We also
provide functions for visual and numerical diagnostics and Bayesian inference
for the fitted models. In this article, we first provide a brief review of the
distributions and techniques used in BAMBI, then describe the capabilities of
the package, and finally conclude with demonstrations of mixture model fitting
using BAMBI on the two real datasets included in the package, one univariate
and one bivariate.
-
This paper considers how to obtain MCMC quantitative convergence bounds which
can be translated into tight complexity bounds in high-dimensional settings. We
propose a modified drift-and-minorization approach, which establishes a
generalized drift condition defined in a subset of the state space. The subset
is called the ``large set'' and is chosen to rule out some ``bad'' states which
have poor drift property when the dimension gets large. Using the ``large set''
together with a ``centered'' drift function, a quantitative bound can be
obtained which can be translated into a tight complexity bound. As a
demonstration, we analyze a certain realistic Gibbs sampler algorithm and
obtain a complexity upper bound for the mixing time, which shows that the
number of iterations required for the Gibbs sampler to converge is constant
under certain conditions on the observed data and the initial state. It is our
hope that this modified drift-and-minorization approach can be employed in many
other specific examples to obtain complexity bounds for high-dimensional Markov
chains.
-
Multiple hypothesis tests are often carried out in practice using p-value
estimates obtained with bootstrap or permutation tests since the analytical
p-values underlying all hypotheses are usually unknown. This article considers
the allocation of a pre-specified total number of Monte Carlo simulations $K
\in \mathbb{N}$ (i.e., permutations or draws from a bootstrap distribution) to
a given number of $m \in \mathbb{N}$ hypotheses in order to approximate their
p-values $p \in [0,1]^m$ in an optimal way, in the sense that the allocation
minimises the total expected number of misclassified hypotheses. A
misclassification occurs if a decision on a single hypothesis, obtained with an
approximated p-value, differs from the one obtained if its p-value was known
analytically. The contribution of this article is threefold: Under the
assumption that $p$ is known and $K \in \mathbb{R}$, and using a normal
approximation of the Binomial distribution, the optimal real-valued allocation
of $K$ simulations to $m$ hypotheses is derived when correcting for
multiplicity with the Bonferroni correction, both when computing the p-value
estimates with or without a pseudo-count. Computational subtleties arising in
the former case will be discussed. Second, with the help of an algorithm based
on simulated annealing, empirical evidence is given that the optimal integer
allocation is likely of the same form as the optimal real-valued allocation,
and that both seem to coincide asympotically. Third, an empirical study on
simulated and real data demonstrates that a recently proposed sampling
algorithm based on Thompson sampling asympotically mimics the optimal
(real-valued) allocation when the p-values are unknown and thus estimated at
runtime.
-
We present a new algorithm which detects the maximal possible number of
matched disjoint pairs satisfying a given caliper when a bipartite matching is
done with respect to a scalar index (e.g., propensity score), and constructs a
corresponding matching. Variable width calipers are compatible with the
technique, provided that the width of the caliper is a Lipschitz function of
the index. If the observations are ordered with respect to the index then the
matching needs $O(N)$ operations, where $N$ is the total number of subjects to
be matched. The case of 1-to-$n$ matching is also considered.
We offer also a new fast algorithm for optimal complete one-to-one matching
on a scalar index when the treatment and control groups are of the same size.
This allows us to improve greedy nearest neighbor matching on a scalar index.
Keywords: propensity score matching, nearest neighbor matching, matching with
caliper, variable width caliper.
-
This tutorial provides a gentle introduction to the particle
Metropolis-Hastings (PMH) algorithm for parameter inference in nonlinear
state-space models together with a software implementation in the statistical
programming language R. We employ a step-by-step approach to develop an
implementation of the PMH algorithm (and the particle filter within) together
with the reader. This final implementation is also available as the package
pmhtutorial in the CRAN repository. Throughout the tutorial, we provide some
intuition as to how the algorithm operates and discuss some solutions to
problems that might occur in practice. To illustrate the use of PMH, we
consider parameter inference in a linear Gaussian state-space model with
synthetic data and a nonlinear stochastic volatility model with real-world
data.
-
Large networks of queueing systems model important real-world systems such as
MapReduce clusters, web-servers, hospitals, call centers and airport passenger
terminals. To model such systems accurately, we must infer queueing parameters
from data. Unfortunately, for many queueing networks there is no clear way to
proceed with parameter inference from data. Approximate Bayesian computation
could offer a straightforward way to infer parameters for such networks if we
could simulate data quickly enough.
We present a computationally efficient method for simulating from a very
general set of queueing networks with the R package queuecomputer. Remarkable
speedups of more than 2 orders of magnitude are observed relative to the
popular DES packages simmer and simpy. We replicate output from these packages
to validate the package.
The package is modular and integrates well with the popular R package dplyr.
Complex queueing networks with tandem, parallel and fork/join topologies can
easily be built with these two packages together. We show how to use this
package with two examples: a call center and an airport terminal.
-
We establish an ordering criterion for the asymptotic variances of two
consistent Markov chain Monte Carlo (MCMC) estimators: an importance sampling
(IS) estimator, based on an approximate reversible chain and subsequent IS
weighting, and a standard MCMC estimator, based on an exact reversible chain.
Essentially, we relax the criterion of the Peskun type covariance ordering by
considering two different invariant probabilities, and obtain, in place of a
strict ordering of asymptotic variances, a bound of the asymptotic variance of
IS by that of the direct MCMC. Simple examples show that IS can have
arbitrarily better or worse asymptotic variance than Metropolis-Hastings and
delayed-acceptance (DA) MCMC. Our ordering implies that IS is guaranteed to be
competitive up to a factor depending on the supremum of the (marginal) IS
weight. We elaborate upon the criterion in case of unbiased estimators as part
of an auxiliary variable framework. We show how the criterion implies
asymptotic variance guarantees for IS in terms of pseudo-marginal (PM) and DA
corrections, essentially if the ratio of exact and approximate likelihoods is
bounded. We also show that convergence of the IS chain can be less affected by
unbounded high-variance unbiased estimators than PM and DA chains.
-
The automation of posterior inference in Bayesian data analysis has enabled
experts and nonexperts alike to use more sophisticated models, engage in faster
exploratory modeling and analysis, and ensure experimental reproducibility.
However, standard automated posterior inference algorithms are not tractable at
the scale of massive modern datasets, and modifications to make them so are
typically model-specific, require expert tuning, and can break theoretical
guarantees on inferential quality. Building on the Bayesian coresets framework,
this work instead takes advantage of data redundancy to shrink the dataset
itself as a preprocessing step, providing fully-automated, scalable Bayesian
inference with theoretical guarantees. We begin with an intuitive reformulation
of Bayesian coreset construction as sparse vector sum approximation, and
demonstrate that its automation and performance-based shortcomings arise from
the use of the supremum norm. To address these shortcomings we develop Hilbert
coresets, i.e., Bayesian coresets constructed under a norm induced by an
inner-product on the log-likelihood function space. We propose two Hilbert
coreset construction algorithms---one based on importance sampling, and one
based on the Frank-Wolfe algorithm---along with theoretical guarantees on
approximation quality as a function of coreset size. Since the exact
computation of the proposed inner-products is model-specific, we automate the
construction with a random finite-dimensional projection of the log-likelihood
functions. The resulting automated coreset construction algorithm is simple to
implement, and experiments on a variety of models with real and synthetic
datasets show that it provides high-quality posterior approximations and a
significant reduction in the computational cost of inference.
-
Bayesian inference for factorial hidden Markov models is challenging due to
the exponentially sized latent variable space. Standard Monte Carlo samplers
can have difficulties effectively exploring the posterior landscape and are
often restricted to exploration around localised regions that depend on
initialisation. We introduce a general purpose ensemble Markov Chain Monte
Carlo (MCMC) technique to improve on existing poorly mixing samplers. This is
achieved by combining parallel tempering and an auxiliary variable scheme to
exchange information between the chains in an efficient way. The latter
exploits a genetic algorithm within an augmented Gibbs sampler. We compare our
technique with various existing samplers in a simulation study as well as in a
cancer genomics application, demonstrating the improvements obtained by our
augmented ensemble approach.
-
Data depth concept offers a variety of powerful and user friendly tools for
robust exploration and inference for multivariate socio-economic phenomena. The
offered techniques may be successfully used in cases of lack of our knowledge
on parametric models generating data due to their nonparametric nature. This
paper presents the R package DepthProc, which is available under GPL-2 licence
on CRAN and R-forge servers for Windows, Linux and OS X platform. The package
consist of among others successful implementations of several data depth
techniques involving multivariate quantile-quantile plots, multivariate scatter
estimators, local Wilcoxon tests for multivariate as well as for functional
data, robust regressions. In order to show the package capabilities, real
datasets concerning United Nations Fourth Millennium Goal and the Internet
users activity are used.
-
Hamiltonian Monte Carlo has emerged as a standard tool for posterior
computation. In this article, we present an extension that can efficiently
explore target distributions with discontinuous densities. Our extension in
particular enables efficient sampling from ordinal parameters though embedding
of probability mass functions into continuous spaces. We motivate our approach
through a theory of discontinuous Hamiltonian dynamics and develop a
corresponding numerical solver. The proposed solver is the first of its kind,
with a remarkable ability to exactly preserve the Hamiltonian. We apply our
algorithm to challenging posterior inference problems to demonstrate its wide
applicability and competitive performance.
-
We consider Bayesian inference for stochastic differential equation mixed
effects models (SDEMEMs) exemplifying tumor response to treatment and regrowth
in mice. We produce an extensive study on how a SDEMEM can be fitted using both
exact inference based on pseudo-marginal MCMC and approximate inference via
Bayesian synthetic likelihoods (BSL). We investigate a two-compartments SDEMEM,
these corresponding to the fractions of tumor cells killed by and survived to a
treatment, respectively. Case study data considers a tumor xenography study
with two treatment groups and one control, each containing 5-8 mice. Results
from the case study and from simulations indicate that the SDEMEM is able to
reproduce the observed growth patterns and that BSL is a robust tool for
inference in SDEMEMs. Finally, we compare the fit of the SDEMEM to a similar
ordinary differential equation model. Due to small sample sizes, strong prior
information is needed to identify all model parameters in the SDEMEM and it
cannot be determined which of the two models is the better in terms of
predicting tumor growth curves. In a simulation study we find that with a
sample of 17 mice per group BSL is able to identify all model parameters and
distinguish treatment groups.
-
Sampling from posterior distributions using Markov chain Monte Carlo (MCMC)
methods can require an exhaustive number of iterations, particularly when the
posterior is multi-modal as the MCMC sampler can become trapped in a local mode
for a large number of iterations. In this paper, we introduce the
pseudo-extended MCMC method as a simple approach for improving the mixing of
the MCMC sampler for multi-modal posterior distributions. The pseudo-extended
method augments the state-space of the posterior using pseudo-samples as
auxiliary variables. On the extended space, the modes of the posterior are
connected, which allows the MCMC sampler to easily move between well-separated
posterior modes. We demonstrate that the pseudo-extended approach delivers
improved MCMC sampling over the Hamiltonian Monte Carlo algorithm on
multi-modal posteriors, including Boltzmann machines and models with
sparsity-inducing priors.
-
A multivariate quantile regression model with a factor structure is proposed
to study data with many responses of interest. The factor structure is allowed
to vary with the quantile levels, which makes our framework more flexible than
the classical factor models. The model is estimated with the nuclear norm
regularization in order to accommodate the high dimensionality of data, but the
incurred optimization problem can only be efficiently solved in an approximate
manner by off-the-shelf optimization methods. Such a scenario is often seen
when the empirical risk is non-smooth or the numerical procedure involves
expensive subroutines such as singular value decomposition. To ensure that the
approximate estimator accurately estimates the model, non-asymptotic bounds on
error of the the approximate estimator is established. For implementation, a
numerical procedure that provably marginalizes the approximate error is
proposed. The merits of our model and the proposed numerical procedures are
demonstrated through Monte Carlo experiments and an application to finance
involving a large pool of asset returns.
-
The method of model averaging has become an important tool to deal with model
uncertainty, for example in situations where a large amount of different
theories exist, as are common in economics. Model averaging is a natural and
formal response to model uncertainty in a Bayesian framework, and most of the
paper deals with Bayesian model averaging. The important role of the prior
assumptions in these Bayesian procedures is highlighted. In addition,
frequentist model averaging methods are also discussed. Numerical methods to
implement these methods are explained, and I point the reader to some freely
available computational resources. The main focus is on uncertainty regarding
the choice of covariates in normal linear regression models, but the paper also
covers other, more challenging, settings, with particular emphasis on sampling
models commonly used in economics. Applications of model averaging in economics
are reviewed and discussed in a wide range of areas, among which growth
economics, production modelling, finance and forecasting macroeconomic
quantities.
-
Gaussian Markov random fields (GMRFs) are popular for modeling dependence in
large areal datasets due to their ease of interpretation and computational
convenience afforded by the sparse precision matrices needed for random
variable generation. Typically in Bayesian computation, GMRFs are updated
jointly in a block Gibbs sampler or componentwise in a single-site sampler via
the full conditional distributions. The former approach can speed convergence
by updating correlated variables all at once, while the latter avoids solving
large matrices. We consider a sampling approach in which the underlying graph
can be cut so that conditionally independent sites are updated simultaneously.
This algorithm allows a practitioner to parallelize updates of subsets of
locations or to take advantage of `vectorized' calculations in a high-level
language such as R. Through both simulated and real data, we demonstrate
computational savings that can be achieved versus both single-site and block
updating, regardless of whether the data are on a regular or an irregular
lattice. The approach provides a good compromise between statistical and
computational efficiency and is accessible to statisticians without expertise
in numerical analysis or advanced computing.
-
Factorial designs with randomization restrictions are often used in
industrial experiments when a complete randomization of trials is impractical.
In the statistics literature, the analysis, construction and isomorphism of
factorial designs has been extensively investigated. Much of the work has been
on a case-by-case basis -- addressing completely randomized designs, randomized
block designs, split-plot designs, etc. separately. In this paper we take a
more unified approach, developing theoretical results and an efficient
relabeling strategy to both construct and check the isomorphism of multi-stage
factorial designs with randomization restrictions. The examples presented in
this paper particularly focus on split-lot designs.
-
Markov chain Monte Carlo methods are a powerful and commonly used family of
numerical methods for sampling from complex probability distributions. As
applications of these methods increase in size and complexity, the need for
efficient methods increases. In this paper, we present a particle ensemble
algorithm. At each iteration, an importance sampling proposal distribution is
formed using an ensemble of particles. A stratified sample is taken from this
distribution and weighted under the posterior, a state-of-the-art ensemble
transport resampling method is then used to create an evenly weighted sample
ready for the next iteration. We demonstrate that this ensemble transport
adaptive importance sampling (ETAIS) method outperforms MCMC methods with
equivalent proposal distributions for low dimensional problems, and in fact
shows better than linear improvements in convergence rates with respect to the
number of ensemble members. We also introduce a new resampling strategy,
multinomial transformation (MT), which while not as accurate as the ensemble
transport resampler, is substantially less costly for large ensemble sizes, and
can then be used in conjunction with ETAIS for complex problems. We also focus
on how algorithmic parameters regarding the mixture proposal can be quickly
tuned to optimise performance. In particular, we demonstrate this methodology's
superior sampling for multimodal problems, such as those arising from inference
for mixture models, and for problems with expensive likelihoods requiring the
solution of a differential equation, for which speed-ups of orders of magnitude
are demonstrated. Likelihood evaluations of the ensemble could be computed in a
distributed manner, suggesting that this methodology is a good candidate for
parallel Bayesian computations.
-
Sequence analysis is being more and more widely used for the analysis of
social sequences and other multivariate categorical time series data. However,
it is often complex to describe, visualize, and compare large sequence data,
especially when there are multiple parallel sequences per subject. Hidden
(latent) Markov models (HMMs) are able to detect underlying latent structures
and they can be used in various longitudinal settings: to account for
measurement error, to detect unobservable states, or to compress information
across several types of observations. Extending to mixture hidden Markov models
(MHMMs) allows clustering data into homogeneous subsets, with or without
external covariates.
The seqHMM package in R is designed for the efficient modeling of sequences
and other categorical time series data containing one or multiple subjects with
one or multiple interdependent sequences using HMMs and MHMMs. Also other
restricted variants of the MHMM can be fitted, e.g., latent class models,
Markov models, mixture Markov models, or even ordinary multinomial regression
models with suitable parameterization of the HMM. Good graphical presentations
of data and models are useful during the whole analysis process from the first
glimpse at the data to model fitting and presentation of results. The package
provides easy options for plotting parallel sequence data, and proposes
visualizing HMMs as directed graphs.
-
We investigate the merits of replication, and provide methods for optimal
design (including replicates), with the goal of obtaining globally accurate
emulation of noisy computer simulation experiments. We first show that
replication can be beneficial from both design and computational perspectives,
in the context of Gaussian process surrogate modeling. We then develop a
lookahead based sequential design scheme that can determine if a new run should
be at an existing input location (i.e., replicate) or at a new one (explore).
When paired with a newly developed heteroskedastic Gaussian process model, our
dynamic design scheme facilitates learning of signal and noise relationships
which can vary throughout the input space. We show that it does so efficiently,
on both computational and statistical grounds. In addition to illustrative
synthetic examples, we demonstrate performance on two challenging real-data
simulation experiments, from inventory management and epidemiology.
-
The development of chemical reaction models aids understanding and prediction
in areas ranging from biology to electrochemistry and combustion. A systematic
approach to building reaction network models uses observational data not only
to estimate unknown parameters, but also to learn model structure. Bayesian
inference provides a natural approach to this data-driven construction of
models. Yet traditional Bayesian model inference methodologies that numerically
evaluate the evidence for each model are often infeasible for nonlinear
reaction network inference, as the number of plausible models can be
combinatorially large. Alternative approaches based on model-space sampling can
enable large-scale network inference, but their realization presents many
challenges. In this paper, we present new computational methods that make
large-scale nonlinear network inference tractable. First, we exploit the
topology of networks describing potential interactions among chemical species
to design improved "between-model" proposals for reversible-jump Markov chain
Monte Carlo. Second, we introduce a sensitivity-based determination of move
types which, when combined with network-aware proposals, yields significant
additional gains in sampling performance. These algorithms are demonstrated on
inference problems drawn from systems biology, with nonlinear differential
equation models of species interactions.





 We propose a general framework for nonasymptotic covariance matrix estimation making use of concentration inequality-based confidence sets. We specify this framework for the estimation of large sparse covariance matrices through incorporation of past thresholding estimators with key emphasis on support recovery. This technique goes beyond past results for thresholding estimators by allowing for a wide range of distributional assumptions beyond merely sub-Gaussian tails. This methodology can furthermore be adapted to a wide range of other estimators and settings. The usage of nonasymptotic dimension-free confidence sets yields good theoretical performance. Through extensive simulations, it is demonstrated to have superior performance when compared with other such methods. In the context of support recovery, we are able to specify a false positive rate and optimize to maximize the true recoveries.
We propose a general framework for nonasymptotic covariance matrix estimation making use of concentration inequality-based confidence sets. We specify this framework for the estimation of large sparse covariance matrices through incorporation of past thresholding estimators with key emphasis on support recovery. This technique goes beyond past results for thresholding estimators by allowing for a wide range of distributional assumptions beyond merely sub-Gaussian tails. This methodology can furthermore be adapted to a wide range of other estimators and settings. The usage of nonasymptotic dimension-free confidence sets yields good theoretical performance. Through extensive simulations, it is demonstrated to have superior performance when compared with other such methods. In the context of support recovery, we are able to specify a false positive rate and optimize to maximize the true recoveries.




 In this article, we present data-subsetting algorithms that allow for the approximate and scalable implementation of the Bayesian bootstrap. They are analogous to two existing algorithms in the frequentist literature: the bag of little bootstraps (Kleiner et al., 2014) and the subsampled double bootstrap (SDB; Sengupta et al., 2016). Our algorithms have appealing theoretical and computational properties that are comparable to those of their frequentist counterparts. Additionally, we provide a strategy for performing lossless inference for a class of functionals of the Bayesian bootstrap, and briefly introduce extensions to the Dirichlet Process.
In this article, we present data-subsetting algorithms that allow for the approximate and scalable implementation of the Bayesian bootstrap. They are analogous to two existing algorithms in the frequentist literature: the bag of little bootstraps (Kleiner et al., 2014) and the subsampled double bootstrap (SDB; Sengupta et al., 2016). Our algorithms have appealing theoretical and computational properties that are comparable to those of their frequentist counterparts. Additionally, we provide a strategy for performing lossless inference for a class of functionals of the Bayesian bootstrap, and briefly introduce extensions to the Dirichlet Process.




 We investigate a new sampling scheme aimed at improving the performance of particle filters whenever (a) there is a significant mismatch between the assumed model dynamics and the actual system, or (b) the posterior probability tends to concentrate in relatively small regions of the state space. The proposed scheme pushes some particles towards specific regions where the likelihood is expected to be high, an operation known as nudging in the geophysics literature. We re-interpret nudging in a form applicable to any particle filtering scheme, as it does not involve any changes in the rest of the algorithm. Since the particles are modified, but the importance weights do not account for this modification, the use of nudging leads to additional bias in the resulting estimators. However, we prove analytically that nudged particle filters can still attain asymptotic convergence with the same error rates as conventional particle methods. Simple analysis also yields an alternative interpretation of the nudging operation that explains its robustness to model errors. Finally, we show numerical results that illustrate the improvements that can be attained using the proposed scheme. In particular, we present nonlinear tracking examples with synthetic data and a model inference example using real-world financial data.
We investigate a new sampling scheme aimed at improving the performance of particle filters whenever (a) there is a significant mismatch between the assumed model dynamics and the actual system, or (b) the posterior probability tends to concentrate in relatively small regions of the state space. The proposed scheme pushes some particles towards specific regions where the likelihood is expected to be high, an operation known as nudging in the geophysics literature. We re-interpret nudging in a form applicable to any particle filtering scheme, as it does not involve any changes in the rest of the algorithm. Since the particles are modified, but the importance weights do not account for this modification, the use of nudging leads to additional bias in the resulting estimators. However, we prove analytically that nudged particle filters can still attain asymptotic convergence with the same error rates as conventional particle methods. Simple analysis also yields an alternative interpretation of the nudging operation that explains its robustness to model errors. Finally, we show numerical results that illustrate the improvements that can be attained using the proposed scheme. In particular, we present nonlinear tracking examples with synthetic data and a model inference example using real-world financial data.




 Statistical analyses of directional or angular data have applications in a variety of fields, such as geology, meteorology and bioinformatics. There is substantial literature on descriptive and inferential techniques for univariate angular data, with the bivariate (or more generally, multivariate) cases receiving more attention in recent years. More specifically, the bivariate wrapped normal, von Mises sine and von Mises cosine distributions, and mixtures thereof, have been proposed for practical use. However, there is a lack of software implementing these distributions and the associated inferential techniques. In this article, we introduce BAMBI, an R package for analyzing bivariate (and univariate) angular data. We implement random data generation, density evaluation, and computation of theoretical summary measures (variances and correlation coefficients) for the three aforementioned bivariate angular distributions, as well as two univariate angular distributions: the univariate wrapped normal and the univariate von Mises distribution. The major contribution of BAMBI to statistical computing is in providing Bayesian methods for modeling angular data using finite mixtures of these distributions. We also provide functions for visual and numerical diagnostics and Bayesian inference for the fitted models. In this article, we first provide a brief review of the distributions and techniques used in BAMBI, then describe the capabilities of the package, and finally conclude with demonstrations of mixture model fitting using BAMBI on the two real datasets included in the package, one univariate and one bivariate.
Statistical analyses of directional or angular data have applications in a variety of fields, such as geology, meteorology and bioinformatics. There is substantial literature on descriptive and inferential techniques for univariate angular data, with the bivariate (or more generally, multivariate) cases receiving more attention in recent years. More specifically, the bivariate wrapped normal, von Mises sine and von Mises cosine distributions, and mixtures thereof, have been proposed for practical use. However, there is a lack of software implementing these distributions and the associated inferential techniques. In this article, we introduce BAMBI, an R package for analyzing bivariate (and univariate) angular data. We implement random data generation, density evaluation, and computation of theoretical summary measures (variances and correlation coefficients) for the three aforementioned bivariate angular distributions, as well as two univariate angular distributions: the univariate wrapped normal and the univariate von Mises distribution. The major contribution of BAMBI to statistical computing is in providing Bayesian methods for modeling angular data using finite mixtures of these distributions. We also provide functions for visual and numerical diagnostics and Bayesian inference for the fitted models. In this article, we first provide a brief review of the distributions and techniques used in BAMBI, then describe the capabilities of the package, and finally conclude with demonstrations of mixture model fitting using BAMBI on the two real datasets included in the package, one univariate and one bivariate.




 This paper considers how to obtain MCMC quantitative convergence bounds which can be translated into tight complexity bounds in high-dimensional settings. We propose a modified drift-and-minorization approach, which establishes a generalized drift condition defined in a subset of the state space. The subset is called the ``large set'' and is chosen to rule out some ``bad'' states which have poor drift property when the dimension gets large. Using the ``large set'' together with a ``centered'' drift function, a quantitative bound can be obtained which can be translated into a tight complexity bound. As a demonstration, we analyze a certain realistic Gibbs sampler algorithm and obtain a complexity upper bound for the mixing time, which shows that the number of iterations required for the Gibbs sampler to converge is constant under certain conditions on the observed data and the initial state. It is our hope that this modified drift-and-minorization approach can be employed in many other specific examples to obtain complexity bounds for high-dimensional Markov chains.
This paper considers how to obtain MCMC quantitative convergence bounds which can be translated into tight complexity bounds in high-dimensional settings. We propose a modified drift-and-minorization approach, which establishes a generalized drift condition defined in a subset of the state space. The subset is called the ``large set'' and is chosen to rule out some ``bad'' states which have poor drift property when the dimension gets large. Using the ``large set'' together with a ``centered'' drift function, a quantitative bound can be obtained which can be translated into a tight complexity bound. As a demonstration, we analyze a certain realistic Gibbs sampler algorithm and obtain a complexity upper bound for the mixing time, which shows that the number of iterations required for the Gibbs sampler to converge is constant under certain conditions on the observed data and the initial state. It is our hope that this modified drift-and-minorization approach can be employed in many other specific examples to obtain complexity bounds for high-dimensional Markov chains.



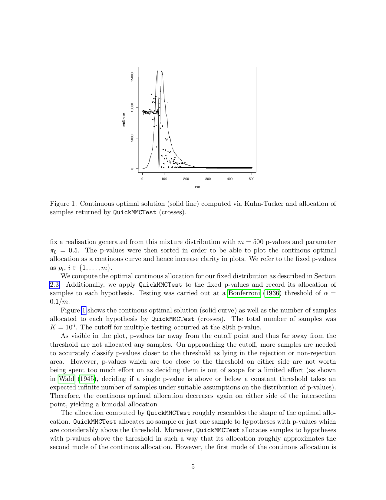
 Multiple hypothesis tests are often carried out in practice using p-value estimates obtained with bootstrap or permutation tests since the analytical p-values underlying all hypotheses are usually unknown. This article considers the allocation of a pre-specified total number of Monte Carlo simulations $K \in \mathbb{N}$ (i.e., permutations or draws from a bootstrap distribution) to a given number of $m \in \mathbb{N}$ hypotheses in order to approximate their p-values $p \in [0,1]^m$ in an optimal way, in the sense that the allocation minimises the total expected number of misclassified hypotheses. A misclassification occurs if a decision on a single hypothesis, obtained with an approximated p-value, differs from the one obtained if its p-value was known analytically. The contribution of this article is threefold: Under the assumption that $p$ is known and $K \in \mathbb{R}$, and using a normal approximation of the Binomial distribution, the optimal real-valued allocation of $K$ simulations to $m$ hypotheses is derived when correcting for multiplicity with the Bonferroni correction, both when computing the p-value estimates with or without a pseudo-count. Computational subtleties arising in the former case will be discussed. Second, with the help of an algorithm based on simulated annealing, empirical evidence is given that the optimal integer allocation is likely of the same form as the optimal real-valued allocation, and that both seem to coincide asympotically. Third, an empirical study on simulated and real data demonstrates that a recently proposed sampling algorithm based on Thompson sampling asympotically mimics the optimal (real-valued) allocation when the p-values are unknown and thus estimated at runtime.
Multiple hypothesis tests are often carried out in practice using p-value estimates obtained with bootstrap or permutation tests since the analytical p-values underlying all hypotheses are usually unknown. This article considers the allocation of a pre-specified total number of Monte Carlo simulations $K \in \mathbb{N}$ (i.e., permutations or draws from a bootstrap distribution) to a given number of $m \in \mathbb{N}$ hypotheses in order to approximate their p-values $p \in [0,1]^m$ in an optimal way, in the sense that the allocation minimises the total expected number of misclassified hypotheses. A misclassification occurs if a decision on a single hypothesis, obtained with an approximated p-value, differs from the one obtained if its p-value was known analytically. The contribution of this article is threefold: Under the assumption that $p$ is known and $K \in \mathbb{R}$, and using a normal approximation of the Binomial distribution, the optimal real-valued allocation of $K$ simulations to $m$ hypotheses is derived when correcting for multiplicity with the Bonferroni correction, both when computing the p-value estimates with or without a pseudo-count. Computational subtleties arising in the former case will be discussed. Second, with the help of an algorithm based on simulated annealing, empirical evidence is given that the optimal integer allocation is likely of the same form as the optimal real-valued allocation, and that both seem to coincide asympotically. Third, an empirical study on simulated and real data demonstrates that a recently proposed sampling algorithm based on Thompson sampling asympotically mimics the optimal (real-valued) allocation when the p-values are unknown and thus estimated at runtime.




 We present a new algorithm which detects the maximal possible number of matched disjoint pairs satisfying a given caliper when a bipartite matching is done with respect to a scalar index (e.g., propensity score), and constructs a corresponding matching. Variable width calipers are compatible with the technique, provided that the width of the caliper is a Lipschitz function of the index. If the observations are ordered with respect to the index then the matching needs $O(N)$ operations, where $N$ is the total number of subjects to be matched. The case of 1-to-$n$ matching is also considered. We offer also a new fast algorithm for optimal complete one-to-one matching on a scalar index when the treatment and control groups are of the same size. This allows us to improve greedy nearest neighbor matching on a scalar index. Keywords: propensity score matching, nearest neighbor matching, matching with caliper, variable width caliper.
We present a new algorithm which detects the maximal possible number of matched disjoint pairs satisfying a given caliper when a bipartite matching is done with respect to a scalar index (e.g., propensity score), and constructs a corresponding matching. Variable width calipers are compatible with the technique, provided that the width of the caliper is a Lipschitz function of the index. If the observations are ordered with respect to the index then the matching needs $O(N)$ operations, where $N$ is the total number of subjects to be matched. The case of 1-to-$n$ matching is also considered. We offer also a new fast algorithm for optimal complete one-to-one matching on a scalar index when the treatment and control groups are of the same size. This allows us to improve greedy nearest neighbor matching on a scalar index. Keywords: propensity score matching, nearest neighbor matching, matching with caliper, variable width caliper.




 This tutorial provides a gentle introduction to the particle Metropolis-Hastings (PMH) algorithm for parameter inference in nonlinear state-space models together with a software implementation in the statistical programming language R. We employ a step-by-step approach to develop an implementation of the PMH algorithm (and the particle filter within) together with the reader. This final implementation is also available as the package pmhtutorial in the CRAN repository. Throughout the tutorial, we provide some intuition as to how the algorithm operates and discuss some solutions to problems that might occur in practice. To illustrate the use of PMH, we consider parameter inference in a linear Gaussian state-space model with synthetic data and a nonlinear stochastic volatility model with real-world data.
This tutorial provides a gentle introduction to the particle Metropolis-Hastings (PMH) algorithm for parameter inference in nonlinear state-space models together with a software implementation in the statistical programming language R. We employ a step-by-step approach to develop an implementation of the PMH algorithm (and the particle filter within) together with the reader. This final implementation is also available as the package pmhtutorial in the CRAN repository. Throughout the tutorial, we provide some intuition as to how the algorithm operates and discuss some solutions to problems that might occur in practice. To illustrate the use of PMH, we consider parameter inference in a linear Gaussian state-space model with synthetic data and a nonlinear stochastic volatility model with real-world data.




 Large networks of queueing systems model important real-world systems such as MapReduce clusters, web-servers, hospitals, call centers and airport passenger terminals. To model such systems accurately, we must infer queueing parameters from data. Unfortunately, for many queueing networks there is no clear way to proceed with parameter inference from data. Approximate Bayesian computation could offer a straightforward way to infer parameters for such networks if we could simulate data quickly enough. We present a computationally efficient method for simulating from a very general set of queueing networks with the R package queuecomputer. Remarkable speedups of more than 2 orders of magnitude are observed relative to the popular DES packages simmer and simpy. We replicate output from these packages to validate the package. The package is modular and integrates well with the popular R package dplyr. Complex queueing networks with tandem, parallel and fork/join topologies can easily be built with these two packages together. We show how to use this package with two examples: a call center and an airport terminal.
Large networks of queueing systems model important real-world systems such as MapReduce clusters, web-servers, hospitals, call centers and airport passenger terminals. To model such systems accurately, we must infer queueing parameters from data. Unfortunately, for many queueing networks there is no clear way to proceed with parameter inference from data. Approximate Bayesian computation could offer a straightforward way to infer parameters for such networks if we could simulate data quickly enough. We present a computationally efficient method for simulating from a very general set of queueing networks with the R package queuecomputer. Remarkable speedups of more than 2 orders of magnitude are observed relative to the popular DES packages simmer and simpy. We replicate output from these packages to validate the package. The package is modular and integrates well with the popular R package dplyr. Complex queueing networks with tandem, parallel and fork/join topologies can easily be built with these two packages together. We show how to use this package with two examples: a call center and an airport terminal.




 We establish an ordering criterion for the asymptotic variances of two consistent Markov chain Monte Carlo (MCMC) estimators: an importance sampling (IS) estimator, based on an approximate reversible chain and subsequent IS weighting, and a standard MCMC estimator, based on an exact reversible chain. Essentially, we relax the criterion of the Peskun type covariance ordering by considering two different invariant probabilities, and obtain, in place of a strict ordering of asymptotic variances, a bound of the asymptotic variance of IS by that of the direct MCMC. Simple examples show that IS can have arbitrarily better or worse asymptotic variance than Metropolis-Hastings and delayed-acceptance (DA) MCMC. Our ordering implies that IS is guaranteed to be competitive up to a factor depending on the supremum of the (marginal) IS weight. We elaborate upon the criterion in case of unbiased estimators as part of an auxiliary variable framework. We show how the criterion implies asymptotic variance guarantees for IS in terms of pseudo-marginal (PM) and DA corrections, essentially if the ratio of exact and approximate likelihoods is bounded. We also show that convergence of the IS chain can be less affected by unbounded high-variance unbiased estimators than PM and DA chains.
We establish an ordering criterion for the asymptotic variances of two consistent Markov chain Monte Carlo (MCMC) estimators: an importance sampling (IS) estimator, based on an approximate reversible chain and subsequent IS weighting, and a standard MCMC estimator, based on an exact reversible chain. Essentially, we relax the criterion of the Peskun type covariance ordering by considering two different invariant probabilities, and obtain, in place of a strict ordering of asymptotic variances, a bound of the asymptotic variance of IS by that of the direct MCMC. Simple examples show that IS can have arbitrarily better or worse asymptotic variance than Metropolis-Hastings and delayed-acceptance (DA) MCMC. Our ordering implies that IS is guaranteed to be competitive up to a factor depending on the supremum of the (marginal) IS weight. We elaborate upon the criterion in case of unbiased estimators as part of an auxiliary variable framework. We show how the criterion implies asymptotic variance guarantees for IS in terms of pseudo-marginal (PM) and DA corrections, essentially if the ratio of exact and approximate likelihoods is bounded. We also show that convergence of the IS chain can be less affected by unbounded high-variance unbiased estimators than PM and DA chains.




 Bayesian inference for factorial hidden Markov models is challenging due to the exponentially sized latent variable space. Standard Monte Carlo samplers can have difficulties effectively exploring the posterior landscape and are often restricted to exploration around localised regions that depend on initialisation. We introduce a general purpose ensemble Markov Chain Monte Carlo (MCMC) technique to improve on existing poorly mixing samplers. This is achieved by combining parallel tempering and an auxiliary variable scheme to exchange information between the chains in an efficient way. The latter exploits a genetic algorithm within an augmented Gibbs sampler. We compare our technique with various existing samplers in a simulation study as well as in a cancer genomics application, demonstrating the improvements obtained by our augmented ensemble approach.
Bayesian inference for factorial hidden Markov models is challenging due to the exponentially sized latent variable space. Standard Monte Carlo samplers can have difficulties effectively exploring the posterior landscape and are often restricted to exploration around localised regions that depend on initialisation. We introduce a general purpose ensemble Markov Chain Monte Carlo (MCMC) technique to improve on existing poorly mixing samplers. This is achieved by combining parallel tempering and an auxiliary variable scheme to exchange information between the chains in an efficient way. The latter exploits a genetic algorithm within an augmented Gibbs sampler. We compare our technique with various existing samplers in a simulation study as well as in a cancer genomics application, demonstrating the improvements obtained by our augmented ensemble approach.



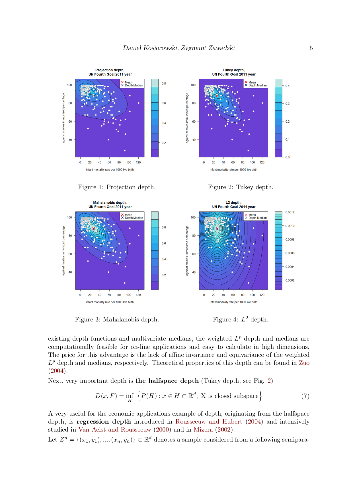
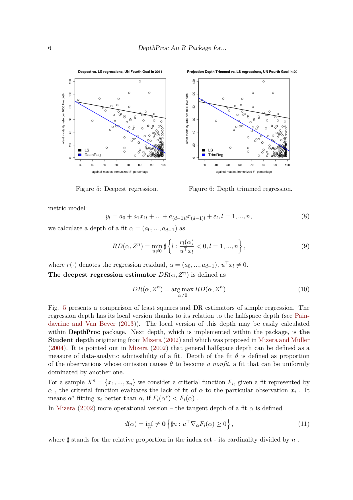 Data depth concept offers a variety of powerful and user friendly tools for robust exploration and inference for multivariate socio-economic phenomena. The offered techniques may be successfully used in cases of lack of our knowledge on parametric models generating data due to their nonparametric nature. This paper presents the R package DepthProc, which is available under GPL-2 licence on CRAN and R-forge servers for Windows, Linux and OS X platform. The package consist of among others successful implementations of several data depth techniques involving multivariate quantile-quantile plots, multivariate scatter estimators, local Wilcoxon tests for multivariate as well as for functional data, robust regressions. In order to show the package capabilities, real datasets concerning United Nations Fourth Millennium Goal and the Internet users activity are used.
Data depth concept offers a variety of powerful and user friendly tools for robust exploration and inference for multivariate socio-economic phenomena. The offered techniques may be successfully used in cases of lack of our knowledge on parametric models generating data due to their nonparametric nature. This paper presents the R package DepthProc, which is available under GPL-2 licence on CRAN and R-forge servers for Windows, Linux and OS X platform. The package consist of among others successful implementations of several data depth techniques involving multivariate quantile-quantile plots, multivariate scatter estimators, local Wilcoxon tests for multivariate as well as for functional data, robust regressions. In order to show the package capabilities, real datasets concerning United Nations Fourth Millennium Goal and the Internet users activity are used.




 Hamiltonian Monte Carlo has emerged as a standard tool for posterior computation. In this article, we present an extension that can efficiently explore target distributions with discontinuous densities. Our extension in particular enables efficient sampling from ordinal parameters though embedding of probability mass functions into continuous spaces. We motivate our approach through a theory of discontinuous Hamiltonian dynamics and develop a corresponding numerical solver. The proposed solver is the first of its kind, with a remarkable ability to exactly preserve the Hamiltonian. We apply our algorithm to challenging posterior inference problems to demonstrate its wide applicability and competitive performance.
Hamiltonian Monte Carlo has emerged as a standard tool for posterior computation. In this article, we present an extension that can efficiently explore target distributions with discontinuous densities. Our extension in particular enables efficient sampling from ordinal parameters though embedding of probability mass functions into continuous spaces. We motivate our approach through a theory of discontinuous Hamiltonian dynamics and develop a corresponding numerical solver. The proposed solver is the first of its kind, with a remarkable ability to exactly preserve the Hamiltonian. We apply our algorithm to challenging posterior inference problems to demonstrate its wide applicability and competitive performance.




 We consider Bayesian inference for stochastic differential equation mixed effects models (SDEMEMs) exemplifying tumor response to treatment and regrowth in mice. We produce an extensive study on how a SDEMEM can be fitted using both exact inference based on pseudo-marginal MCMC and approximate inference via Bayesian synthetic likelihoods (BSL). We investigate a two-compartments SDEMEM, these corresponding to the fractions of tumor cells killed by and survived to a treatment, respectively. Case study data considers a tumor xenography study with two treatment groups and one control, each containing 5-8 mice. Results from the case study and from simulations indicate that the SDEMEM is able to reproduce the observed growth patterns and that BSL is a robust tool for inference in SDEMEMs. Finally, we compare the fit of the SDEMEM to a similar ordinary differential equation model. Due to small sample sizes, strong prior information is needed to identify all model parameters in the SDEMEM and it cannot be determined which of the two models is the better in terms of predicting tumor growth curves. In a simulation study we find that with a sample of 17 mice per group BSL is able to identify all model parameters and distinguish treatment groups.
We consider Bayesian inference for stochastic differential equation mixed effects models (SDEMEMs) exemplifying tumor response to treatment and regrowth in mice. We produce an extensive study on how a SDEMEM can be fitted using both exact inference based on pseudo-marginal MCMC and approximate inference via Bayesian synthetic likelihoods (BSL). We investigate a two-compartments SDEMEM, these corresponding to the fractions of tumor cells killed by and survived to a treatment, respectively. Case study data considers a tumor xenography study with two treatment groups and one control, each containing 5-8 mice. Results from the case study and from simulations indicate that the SDEMEM is able to reproduce the observed growth patterns and that BSL is a robust tool for inference in SDEMEMs. Finally, we compare the fit of the SDEMEM to a similar ordinary differential equation model. Due to small sample sizes, strong prior information is needed to identify all model parameters in the SDEMEM and it cannot be determined which of the two models is the better in terms of predicting tumor growth curves. In a simulation study we find that with a sample of 17 mice per group BSL is able to identify all model parameters and distinguish treatment groups.




 Sampling from posterior distributions using Markov chain Monte Carlo (MCMC) methods can require an exhaustive number of iterations, particularly when the posterior is multi-modal as the MCMC sampler can become trapped in a local mode for a large number of iterations. In this paper, we introduce the pseudo-extended MCMC method as a simple approach for improving the mixing of the MCMC sampler for multi-modal posterior distributions. The pseudo-extended method augments the state-space of the posterior using pseudo-samples as auxiliary variables. On the extended space, the modes of the posterior are connected, which allows the MCMC sampler to easily move between well-separated posterior modes. We demonstrate that the pseudo-extended approach delivers improved MCMC sampling over the Hamiltonian Monte Carlo algorithm on multi-modal posteriors, including Boltzmann machines and models with sparsity-inducing priors.
Sampling from posterior distributions using Markov chain Monte Carlo (MCMC) methods can require an exhaustive number of iterations, particularly when the posterior is multi-modal as the MCMC sampler can become trapped in a local mode for a large number of iterations. In this paper, we introduce the pseudo-extended MCMC method as a simple approach for improving the mixing of the MCMC sampler for multi-modal posterior distributions. The pseudo-extended method augments the state-space of the posterior using pseudo-samples as auxiliary variables. On the extended space, the modes of the posterior are connected, which allows the MCMC sampler to easily move between well-separated posterior modes. We demonstrate that the pseudo-extended approach delivers improved MCMC sampling over the Hamiltonian Monte Carlo algorithm on multi-modal posteriors, including Boltzmann machines and models with sparsity-inducing priors.

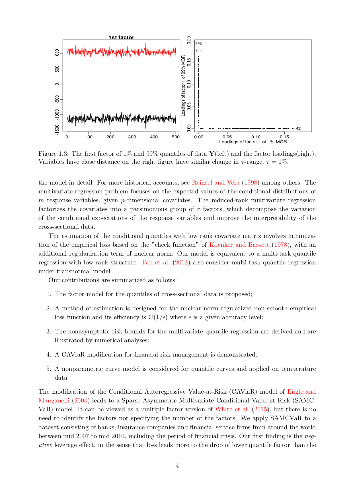

 A multivariate quantile regression model with a factor structure is proposed to study data with many responses of interest. The factor structure is allowed to vary with the quantile levels, which makes our framework more flexible than the classical factor models. The model is estimated with the nuclear norm regularization in order to accommodate the high dimensionality of data, but the incurred optimization problem can only be efficiently solved in an approximate manner by off-the-shelf optimization methods. Such a scenario is often seen when the empirical risk is non-smooth or the numerical procedure involves expensive subroutines such as singular value decomposition. To ensure that the approximate estimator accurately estimates the model, non-asymptotic bounds on error of the the approximate estimator is established. For implementation, a numerical procedure that provably marginalizes the approximate error is proposed. The merits of our model and the proposed numerical procedures are demonstrated through Monte Carlo experiments and an application to finance involving a large pool of asset returns.
A multivariate quantile regression model with a factor structure is proposed to study data with many responses of interest. The factor structure is allowed to vary with the quantile levels, which makes our framework more flexible than the classical factor models. The model is estimated with the nuclear norm regularization in order to accommodate the high dimensionality of data, but the incurred optimization problem can only be efficiently solved in an approximate manner by off-the-shelf optimization methods. Such a scenario is often seen when the empirical risk is non-smooth or the numerical procedure involves expensive subroutines such as singular value decomposition. To ensure that the approximate estimator accurately estimates the model, non-asymptotic bounds on error of the the approximate estimator is established. For implementation, a numerical procedure that provably marginalizes the approximate error is proposed. The merits of our model and the proposed numerical procedures are demonstrated through Monte Carlo experiments and an application to finance involving a large pool of asset returns.




 Gaussian Markov random fields (GMRFs) are popular for modeling dependence in large areal datasets due to their ease of interpretation and computational convenience afforded by the sparse precision matrices needed for random variable generation. Typically in Bayesian computation, GMRFs are updated jointly in a block Gibbs sampler or componentwise in a single-site sampler via the full conditional distributions. The former approach can speed convergence by updating correlated variables all at once, while the latter avoids solving large matrices. We consider a sampling approach in which the underlying graph can be cut so that conditionally independent sites are updated simultaneously. This algorithm allows a practitioner to parallelize updates of subsets of locations or to take advantage of `vectorized' calculations in a high-level language such as R. Through both simulated and real data, we demonstrate computational savings that can be achieved versus both single-site and block updating, regardless of whether the data are on a regular or an irregular lattice. The approach provides a good compromise between statistical and computational efficiency and is accessible to statisticians without expertise in numerical analysis or advanced computing.
Gaussian Markov random fields (GMRFs) are popular for modeling dependence in large areal datasets due to their ease of interpretation and computational convenience afforded by the sparse precision matrices needed for random variable generation. Typically in Bayesian computation, GMRFs are updated jointly in a block Gibbs sampler or componentwise in a single-site sampler via the full conditional distributions. The former approach can speed convergence by updating correlated variables all at once, while the latter avoids solving large matrices. We consider a sampling approach in which the underlying graph can be cut so that conditionally independent sites are updated simultaneously. This algorithm allows a practitioner to parallelize updates of subsets of locations or to take advantage of `vectorized' calculations in a high-level language such as R. Through both simulated and real data, we demonstrate computational savings that can be achieved versus both single-site and block updating, regardless of whether the data are on a regular or an irregular lattice. The approach provides a good compromise between statistical and computational efficiency and is accessible to statisticians without expertise in numerical analysis or advanced computing.




 Factorial designs with randomization restrictions are often used in industrial experiments when a complete randomization of trials is impractical. In the statistics literature, the analysis, construction and isomorphism of factorial designs has been extensively investigated. Much of the work has been on a case-by-case basis -- addressing completely randomized designs, randomized block designs, split-plot designs, etc. separately. In this paper we take a more unified approach, developing theoretical results and an efficient relabeling strategy to both construct and check the isomorphism of multi-stage factorial designs with randomization restrictions. The examples presented in this paper particularly focus on split-lot designs.
Factorial designs with randomization restrictions are often used in industrial experiments when a complete randomization of trials is impractical. In the statistics literature, the analysis, construction and isomorphism of factorial designs has been extensively investigated. Much of the work has been on a case-by-case basis -- addressing completely randomized designs, randomized block designs, split-plot designs, etc. separately. In this paper we take a more unified approach, developing theoretical results and an efficient relabeling strategy to both construct and check the isomorphism of multi-stage factorial designs with randomization restrictions. The examples presented in this paper particularly focus on split-lot designs.




 Markov chain Monte Carlo methods are a powerful and commonly used family of numerical methods for sampling from complex probability distributions. As applications of these methods increase in size and complexity, the need for efficient methods increases. In this paper, we present a particle ensemble algorithm. At each iteration, an importance sampling proposal distribution is formed using an ensemble of particles. A stratified sample is taken from this distribution and weighted under the posterior, a state-of-the-art ensemble transport resampling method is then used to create an evenly weighted sample ready for the next iteration. We demonstrate that this ensemble transport adaptive importance sampling (ETAIS) method outperforms MCMC methods with equivalent proposal distributions for low dimensional problems, and in fact shows better than linear improvements in convergence rates with respect to the number of ensemble members. We also introduce a new resampling strategy, multinomial transformation (MT), which while not as accurate as the ensemble transport resampler, is substantially less costly for large ensemble sizes, and can then be used in conjunction with ETAIS for complex problems. We also focus on how algorithmic parameters regarding the mixture proposal can be quickly tuned to optimise performance. In particular, we demonstrate this methodology's superior sampling for multimodal problems, such as those arising from inference for mixture models, and for problems with expensive likelihoods requiring the solution of a differential equation, for which speed-ups of orders of magnitude are demonstrated. Likelihood evaluations of the ensemble could be computed in a distributed manner, suggesting that this methodology is a good candidate for parallel Bayesian computations.
Markov chain Monte Carlo methods are a powerful and commonly used family of numerical methods for sampling from complex probability distributions. As applications of these methods increase in size and complexity, the need for efficient methods increases. In this paper, we present a particle ensemble algorithm. At each iteration, an importance sampling proposal distribution is formed using an ensemble of particles. A stratified sample is taken from this distribution and weighted under the posterior, a state-of-the-art ensemble transport resampling method is then used to create an evenly weighted sample ready for the next iteration. We demonstrate that this ensemble transport adaptive importance sampling (ETAIS) method outperforms MCMC methods with equivalent proposal distributions for low dimensional problems, and in fact shows better than linear improvements in convergence rates with respect to the number of ensemble members. We also introduce a new resampling strategy, multinomial transformation (MT), which while not as accurate as the ensemble transport resampler, is substantially less costly for large ensemble sizes, and can then be used in conjunction with ETAIS for complex problems. We also focus on how algorithmic parameters regarding the mixture proposal can be quickly tuned to optimise performance. In particular, we demonstrate this methodology's superior sampling for multimodal problems, such as those arising from inference for mixture models, and for problems with expensive likelihoods requiring the solution of a differential equation, for which speed-ups of orders of magnitude are demonstrated. Likelihood evaluations of the ensemble could be computed in a distributed manner, suggesting that this methodology is a good candidate for parallel Bayesian computations.




 Sequence analysis is being more and more widely used for the analysis of social sequences and other multivariate categorical time series data. However, it is often complex to describe, visualize, and compare large sequence data, especially when there are multiple parallel sequences per subject. Hidden (latent) Markov models (HMMs) are able to detect underlying latent structures and they can be used in various longitudinal settings: to account for measurement error, to detect unobservable states, or to compress information across several types of observations. Extending to mixture hidden Markov models (MHMMs) allows clustering data into homogeneous subsets, with or without external covariates. The seqHMM package in R is designed for the efficient modeling of sequences and other categorical time series data containing one or multiple subjects with one or multiple interdependent sequences using HMMs and MHMMs. Also other restricted variants of the MHMM can be fitted, e.g., latent class models, Markov models, mixture Markov models, or even ordinary multinomial regression models with suitable parameterization of the HMM. Good graphical presentations of data and models are useful during the whole analysis process from the first glimpse at the data to model fitting and presentation of results. The package provides easy options for plotting parallel sequence data, and proposes visualizing HMMs as directed graphs.
Sequence analysis is being more and more widely used for the analysis of social sequences and other multivariate categorical time series data. However, it is often complex to describe, visualize, and compare large sequence data, especially when there are multiple parallel sequences per subject. Hidden (latent) Markov models (HMMs) are able to detect underlying latent structures and they can be used in various longitudinal settings: to account for measurement error, to detect unobservable states, or to compress information across several types of observations. Extending to mixture hidden Markov models (MHMMs) allows clustering data into homogeneous subsets, with or without external covariates. The seqHMM package in R is designed for the efficient modeling of sequences and other categorical time series data containing one or multiple subjects with one or multiple interdependent sequences using HMMs and MHMMs. Also other restricted variants of the MHMM can be fitted, e.g., latent class models, Markov models, mixture Markov models, or even ordinary multinomial regression models with suitable parameterization of the HMM. Good graphical presentations of data and models are useful during the whole analysis process from the first glimpse at the data to model fitting and presentation of results. The package provides easy options for plotting parallel sequence data, and proposes visualizing HMMs as directed graphs.



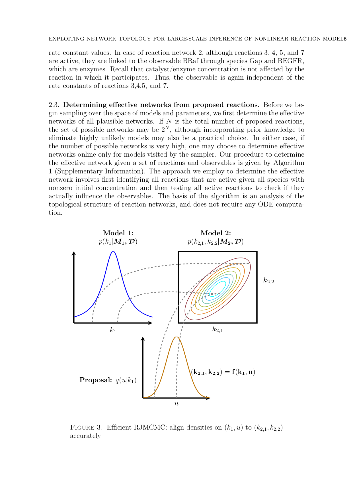
 The development of chemical reaction models aids understanding and prediction in areas ranging from biology to electrochemistry and combustion. A systematic approach to building reaction network models uses observational data not only to estimate unknown parameters, but also to learn model structure. Bayesian inference provides a natural approach to this data-driven construction of models. Yet traditional Bayesian model inference methodologies that numerically evaluate the evidence for each model are often infeasible for nonlinear reaction network inference, as the number of plausible models can be combinatorially large. Alternative approaches based on model-space sampling can enable large-scale network inference, but their realization presents many challenges. In this paper, we present new computational methods that make large-scale nonlinear network inference tractable. First, we exploit the topology of networks describing potential interactions among chemical species to design improved "between-model" proposals for reversible-jump Markov chain Monte Carlo. Second, we introduce a sensitivity-based determination of move types which, when combined with network-aware proposals, yields significant additional gains in sampling performance. These algorithms are demonstrated on inference problems drawn from systems biology, with nonlinear differential equation models of species interactions.
The development of chemical reaction models aids understanding and prediction in areas ranging from biology to electrochemistry and combustion. A systematic approach to building reaction network models uses observational data not only to estimate unknown parameters, but also to learn model structure. Bayesian inference provides a natural approach to this data-driven construction of models. Yet traditional Bayesian model inference methodologies that numerically evaluate the evidence for each model are often infeasible for nonlinear reaction network inference, as the number of plausible models can be combinatorially large. Alternative approaches based on model-space sampling can enable large-scale network inference, but their realization presents many challenges. In this paper, we present new computational methods that make large-scale nonlinear network inference tractable. First, we exploit the topology of networks describing potential interactions among chemical species to design improved "between-model" proposals for reversible-jump Markov chain Monte Carlo. Second, we introduce a sensitivity-based determination of move types which, when combined with network-aware proposals, yields significant additional gains in sampling performance. These algorithms are demonstrated on inference problems drawn from systems biology, with nonlinear differential equation models of species interactions.